LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_1.5wLII_02657_1
Total number of 3D structures: 37
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
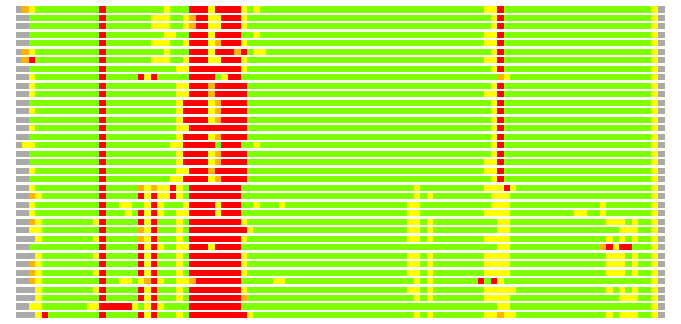
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1cm7_A |
363 |
101 |
90 |
1.10 |
20.00 |
88.015 |
7.524 |
T P |
| 1ayq_B |
357 |
101 |
91 |
1.28 |
12.09 |
87.263 |
6.601 |
T P |
| 2ayq_B |
357 |
101 |
91 |
1.28 |
12.09 |
87.263 |
6.601 |
T P |
| 1a05_A |
357 |
101 |
89 |
1.05 |
19.10 |
86.586 |
7.710 |
T P |
| 1v53_A |
356 |
101 |
90 |
1.23 |
12.22 |
86.434 |
6.769 |
T P |
| 1cnz_A |
363 |
101 |
90 |
1.26 |
20.00 |
86.377 |
6.609 |
T P |
| 1v5b_B |
357 |
101 |
90 |
1.26 |
12.22 |
85.991 |
6.619 |
T P |
| 1xac_A |
345 |
101 |
88 |
0.95 |
18.18 |
85.683 |
8.342 |
T P |
| 1w0d_A |
337 |
101 |
89 |
1.28 |
15.73 |
85.652 |
6.458 |
T P |
| 1wal_A |
345 |
101 |
88 |
1.05 |
18.18 |
85.361 |
7.630 |
T P |
| 1gc9_A |
345 |
101 |
88 |
1.05 |
18.18 |
85.361 |
7.626 |
T P |
| 1idm_A |
343 |
101 |
88 |
1.05 |
18.18 |
85.340 |
7.630 |
T P |
| 1ipd_A |
345 |
101 |
88 |
1.05 |
18.18 |
85.340 |
7.655 |
T P |
| 1dpz_A |
345 |
101 |
88 |
1.07 |
18.18 |
85.318 |
7.544 |
T P |
| 1dr0_A |
346 |
101 |
87 |
0.95 |
18.39 |
85.318 |
8.300 |
T P |
| 1g2u_A |
345 |
101 |
88 |
1.06 |
18.18 |
85.261 |
7.562 |
T P |
| 1vlc_A |
362 |
101 |
89 |
1.12 |
15.73 |
85.218 |
7.315 |
T P |
| 1xaa_A |
345 |
101 |
88 |
1.06 |
18.18 |
85.183 |
7.557 |
T P |
| 1osj_A |
345 |
101 |
88 |
1.09 |
18.18 |
85.070 |
7.366 |
T P |
| 1gc8_A |
345 |
101 |
88 |
1.12 |
18.18 |
84.962 |
7.185 |
T P |
| 1dr8_A |
344 |
101 |
88 |
1.12 |
18.18 |
84.914 |
7.199 |
T P |
| 2d1c_A |
495 |
101 |
87 |
1.62 |
13.79 |
82.413 |
5.053 |
T P |
| 3blv_H |
348 |
101 |
86 |
1.46 |
9.30 |
81.913 |
5.496 |
T P |
| 3blx_E |
341 |
101 |
89 |
1.53 |
13.48 |
81.666 |
5.446 |
T P |
| 3blv_C |
344 |
101 |
88 |
1.52 |
13.64 |
81.562 |
5.446 |
T P |
| 6icd_A |
414 |
101 |
87 |
1.59 |
8.05 |
81.258 |
5.150 |
T P |
| 2e0c_A |
401 |
101 |
87 |
1.56 |
14.94 |
81.136 |
5.230 |
T P |
| 1pb1_A |
416 |
101 |
87 |
1.60 |
6.90 |
80.984 |
5.119 |
T P |
| 1wpw_A |
336 |
101 |
85 |
1.37 |
11.76 |
80.958 |
5.766 |
T P |
| 1cw7_A |
415 |
101 |
86 |
1.54 |
6.98 |
80.932 |
5.257 |
T P |
| 1hj6_A |
414 |
101 |
87 |
1.61 |
8.05 |
80.928 |
5.081 |
T P |
| 1bl5_A |
414 |
101 |
87 |
1.60 |
8.05 |
80.919 |
5.125 |
T P |
| 3blx_B |
346 |
101 |
87 |
1.63 |
9.20 |
80.862 |
5.038 |
T P |
| 1iso_A |
414 |
101 |
86 |
1.54 |
6.98 |
80.728 |
5.248 |
T P |
| 1hqs_A |
423 |
101 |
86 |
1.55 |
9.30 |
80.376 |
5.204 |
T P |
| 1x0l_A |
333 |
101 |
84 |
1.34 |
9.52 |
80.259 |
5.824 |
T P |
| 2d4v_A |
427 |
101 |
84 |
1.61 |
9.52 |
78.999 |
4.921 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]