LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_102.5wLII_11068_84
Total number of 3D structures: 74
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
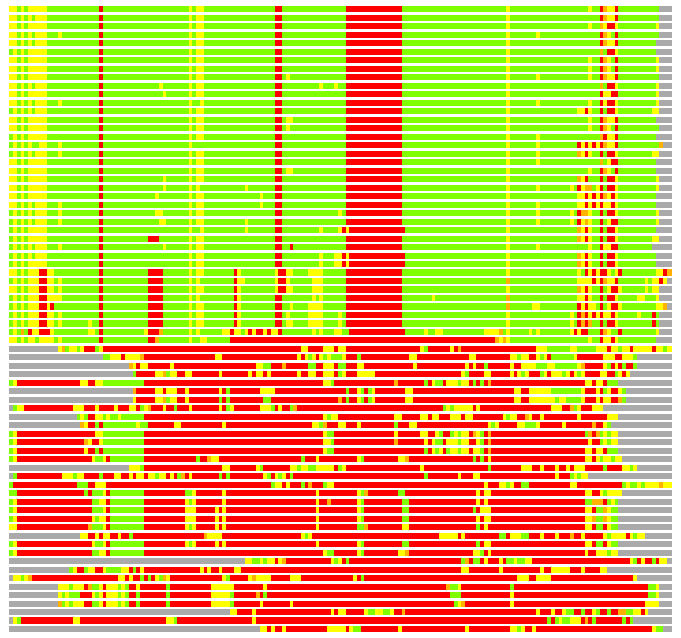
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ynd_A |
164 |
177 |
154 |
1.10 |
18.83 |
84.074 |
12.810 |
T P |
| 2z6w_A |
164 |
177 |
154 |
1.13 |
20.13 |
83.760 |
12.473 |
T P |
| 2alf_A |
164 |
177 |
154 |
1.16 |
18.83 |
83.750 |
12.218 |
T P |
| 2bit_X |
164 |
177 |
154 |
1.18 |
20.13 |
83.584 |
12.002 |
T P |
| 2cfe_A |
162 |
177 |
153 |
1.17 |
20.26 |
83.249 |
12.016 |
T P |
| 1qnh_B |
170 |
177 |
153 |
1.16 |
18.95 |
83.244 |
12.104 |
T P |
| 1zcx_A |
164 |
177 |
154 |
1.23 |
16.88 |
83.140 |
11.571 |
T P |
| 2r99_A |
164 |
177 |
154 |
1.23 |
16.88 |
83.140 |
11.571 |
T P |
| 1awq_A |
164 |
177 |
154 |
1.22 |
18.83 |
83.118 |
11.671 |
T P |
| 2hqj_A |
179 |
177 |
154 |
1.24 |
16.23 |
83.043 |
11.452 |
T P |
| 2igv_A |
172 |
177 |
153 |
1.19 |
18.95 |
83.036 |
11.889 |
T P |
| 2cmt_A |
164 |
177 |
153 |
1.19 |
18.95 |
82.991 |
11.892 |
T P |
| 1cyn_A |
178 |
177 |
152 |
1.21 |
23.03 |
82.984 |
11.643 |
T P |
| 1m9e_A |
164 |
177 |
153 |
1.19 |
18.95 |
82.967 |
11.865 |
T P |
| 1aws_A |
164 |
177 |
154 |
1.24 |
18.18 |
82.957 |
11.492 |
T P |
| 1vdn_A |
161 |
177 |
153 |
1.20 |
17.65 |
82.910 |
11.788 |
T P |
| 2esl_A |
181 |
177 |
152 |
1.33 |
21.71 |
82.870 |
10.637 |
T P |
| 2rmc_A |
182 |
177 |
152 |
1.24 |
21.71 |
82.870 |
11.340 |
T P |
| 1qng_A |
170 |
177 |
153 |
1.22 |
19.61 |
82.632 |
11.590 |
T P |
| 1zmf_A |
162 |
177 |
153 |
1.25 |
16.99 |
82.488 |
11.373 |
T P |
| 1ihg_A |
364 |
177 |
152 |
1.22 |
19.08 |
82.186 |
11.509 |
T P |
| 2gw2_A |
173 |
177 |
152 |
1.36 |
15.79 |
81.856 |
10.395 |
T P |
| 1mzw_A |
173 |
177 |
151 |
1.34 |
17.88 |
81.728 |
10.481 |
T P |
| 1z81_A |
186 |
177 |
151 |
1.33 |
18.54 |
81.586 |
10.596 |
T P |
| 1a58_A |
177 |
177 |
152 |
1.41 |
18.42 |
81.353 |
10.092 |
T P |
| 2he9_A |
172 |
177 |
152 |
1.38 |
16.45 |
81.306 |
10.287 |
T P |
| 1xo7_A |
166 |
177 |
150 |
1.26 |
20.00 |
81.284 |
11.048 |
T P |
| 1h0p_A |
182 |
177 |
149 |
1.24 |
18.79 |
81.257 |
11.110 |
T P |
| 2poy_B |
172 |
177 |
150 |
1.23 |
19.33 |
81.180 |
11.246 |
T P |
| 2haq_A |
166 |
177 |
150 |
1.29 |
19.33 |
81.029 |
10.794 |
T P |
| 3bt8_A |
166 |
177 |
150 |
1.30 |
19.33 |
80.801 |
10.703 |
T P |
| 1zkc_A |
187 |
177 |
146 |
1.52 |
15.75 |
78.048 |
9.002 |
T P |
| 2ok3_A |
160 |
177 |
147 |
1.60 |
15.65 |
77.416 |
8.633 |
T P |
| 2oju_B |
166 |
177 |
145 |
1.48 |
16.55 |
77.079 |
9.184 |
T P |
| 2poe_A |
162 |
177 |
145 |
1.47 |
16.55 |
76.742 |
9.235 |
T P |
| 2hq6_A |
170 |
177 |
145 |
1.61 |
17.24 |
76.452 |
8.493 |
T P |
| 2a2n_C |
165 |
177 |
143 |
1.56 |
15.38 |
75.280 |
8.606 |
T P |
| 2fu0_A |
155 |
177 |
143 |
1.52 |
16.78 |
75.183 |
8.828 |
T P |
| 1xwn_A |
166 |
177 |
140 |
1.94 |
14.29 |
66.049 |
6.872 |
T P |
| 2c3b_A |
141 |
177 |
97 |
1.84 |
11.34 |
50.651 |
4.999 |
T P |
| 1nm8_A |
591 |
177 |
53 |
2.68 |
7.55 |
19.800 |
1.910 |
T P |
| 2v2b_A |
272 |
177 |
50 |
2.64 |
8.00 |
18.999 |
1.824 |
T P |
| 1gt7_A |
274 |
177 |
46 |
2.52 |
2.17 |
18.526 |
1.758 |
T P |
| 2v9n_A |
274 |
177 |
50 |
2.78 |
6.00 |
18.335 |
1.738 |
T P |
| 1dzz_P |
208 |
177 |
41 |
2.38 |
14.63 |
16.538 |
1.656 |
T P |
| 2v9l_A |
274 |
177 |
42 |
2.65 |
4.76 |
16.433 |
1.528 |
T P |
| 1ojr_A |
274 |
177 |
42 |
2.54 |
4.76 |
16.404 |
1.590 |
T P |
| 2zco_A |
284 |
177 |
44 |
2.89 |
4.55 |
16.379 |
1.472 |
T P |
| 2opi_B |
209 |
177 |
42 |
2.76 |
7.14 |
16.164 |
1.466 |
T P |
| 2v9g_B |
274 |
177 |
41 |
2.53 |
9.76 |
16.152 |
1.559 |
T P |
| 1e48_P |
206 |
177 |
40 |
2.50 |
10.00 |
16.081 |
1.541 |
T P |
| 1fua_A |
206 |
177 |
39 |
2.47 |
7.69 |
15.967 |
1.519 |
T P |
| 1e4c_P |
206 |
177 |
39 |
2.31 |
10.26 |
15.688 |
1.616 |
T P |
| 1e49_P |
206 |
177 |
38 |
2.35 |
5.26 |
15.425 |
1.549 |
T P |
| 2v9e_A |
274 |
177 |
38 |
2.81 |
7.89 |
15.336 |
1.307 |
T P |
| 1ezf_B |
324 |
177 |
40 |
2.64 |
2.50 |
15.314 |
1.458 |
T P |
| 2z7b_A |
237 |
177 |
38 |
2.49 |
2.63 |
15.056 |
1.468 |
T P |
| 1e4b_P |
206 |
177 |
37 |
2.37 |
2.70 |
14.997 |
1.499 |
T P |
| 1e4a_P |
205 |
177 |
35 |
2.32 |
5.71 |
14.954 |
1.445 |
T P |
| 1e47_P |
206 |
177 |
37 |
2.28 |
5.41 |
14.864 |
1.557 |
T P |
| 1dzu_P |
209 |
177 |
37 |
2.38 |
5.41 |
14.817 |
1.494 |
T P |
| 1dzx_P |
208 |
177 |
36 |
2.41 |
5.56 |
14.784 |
1.437 |
T P |
| 1jdi_A |
223 |
177 |
42 |
2.91 |
2.38 |
14.733 |
1.397 |
T P |
| 1dzv_P |
206 |
177 |
35 |
2.16 |
5.71 |
14.636 |
1.546 |
T P |
| 1e46_P |
206 |
177 |
34 |
2.42 |
2.94 |
13.818 |
1.348 |
T P |
| 1pvt_A |
232 |
177 |
33 |
2.50 |
0.00 |
13.460 |
1.269 |
T P |
| 1k0w_A |
223 |
177 |
31 |
2.82 |
6.45 |
13.160 |
1.062 |
T P |
| 2v2a_A |
274 |
177 |
35 |
2.82 |
8.57 |
12.897 |
1.197 |
T P |
| 2v9m_A |
274 |
177 |
34 |
2.63 |
2.94 |
12.854 |
1.245 |
T P |
| 2v9i_A |
272 |
177 |
32 |
2.36 |
3.12 |
12.790 |
1.302 |
T P |
| 2v29_A |
274 |
177 |
34 |
2.79 |
5.88 |
12.147 |
1.175 |
T P |
| 2fk5_A |
195 |
177 |
31 |
2.69 |
12.90 |
11.879 |
1.110 |
T P |
| 2irp_A |
206 |
177 |
20 |
2.42 |
10.00 |
8.990 |
0.793 |
T P |
| 1dzw_P |
206 |
177 |
22 |
2.78 |
0.00 |
8.893 |
0.765 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]