LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_104.5wLII_11068_86
Total number of 3D structures: 54
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
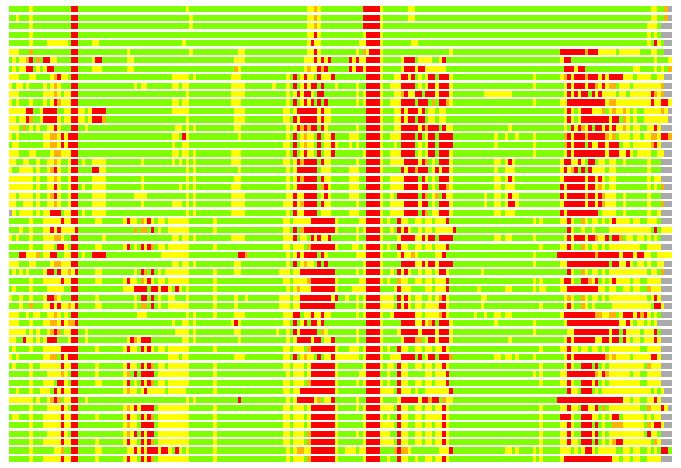
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1z5o_A |
235 |
191 |
183 |
1.05 |
14.75 |
93.786 |
15.880 |
T P |
| 1z5n_A |
234 |
191 |
183 |
1.07 |
15.30 |
93.554 |
15.627 |
T P |
| 3dp9_C |
230 |
191 |
181 |
0.92 |
17.68 |
93.217 |
17.815 |
T P |
| 3bl6_A |
230 |
191 |
181 |
1.03 |
15.47 |
92.695 |
16.042 |
T P |
| 1zos_A |
230 |
191 |
181 |
1.31 |
12.15 |
90.654 |
12.799 |
T P |
| 1jys_A |
226 |
191 |
173 |
1.59 |
16.18 |
84.548 |
10.236 |
T P |
| 2h8g_A |
246 |
191 |
169 |
1.66 |
12.43 |
82.275 |
9.586 |
T P |
| 3bsf_A |
244 |
191 |
166 |
1.77 |
11.45 |
74.844 |
8.857 |
T P |
| 1ecp_A |
237 |
191 |
158 |
1.87 |
10.13 |
71.563 |
8.041 |
T P |
| 1vhw_A |
237 |
191 |
162 |
2.01 |
10.49 |
71.421 |
7.666 |
T P |
| 1odk_B |
234 |
191 |
162 |
1.95 |
15.43 |
70.200 |
7.893 |
T P |
| 1pk9_A |
237 |
191 |
156 |
1.92 |
9.62 |
70.093 |
7.716 |
T P |
| 2guw_B |
418 |
191 |
158 |
1.93 |
16.46 |
69.956 |
7.770 |
T P |
| 1t8s_B |
444 |
191 |
153 |
1.82 |
16.99 |
69.430 |
7.976 |
T P |
| 1q1g_A |
243 |
191 |
158 |
1.95 |
9.49 |
69.353 |
7.690 |
T P |
| 2isc_D |
238 |
191 |
157 |
1.98 |
11.46 |
68.933 |
7.553 |
T P |
| 2i4t_B |
235 |
191 |
156 |
1.93 |
12.82 |
68.818 |
7.672 |
T P |
| 1pw7_A |
237 |
191 |
156 |
1.89 |
9.62 |
68.649 |
7.847 |
T P |
| 1u1g_B |
251 |
191 |
162 |
1.98 |
12.35 |
68.567 |
7.802 |
T P |
| 2hrd_E |
252 |
191 |
160 |
1.90 |
11.88 |
68.227 |
7.982 |
T P |
| 2qdk_A |
252 |
191 |
158 |
1.96 |
12.66 |
68.056 |
7.652 |
T P |
| 3c74_D |
251 |
191 |
159 |
1.99 |
12.58 |
67.726 |
7.591 |
T P |
| 1lx7_A |
250 |
191 |
157 |
2.03 |
12.10 |
66.764 |
7.370 |
T P |
| 1rxy_A |
250 |
191 |
157 |
2.01 |
12.74 |
66.626 |
7.431 |
T P |
| 2oxf_A |
250 |
191 |
155 |
1.95 |
12.90 |
66.177 |
7.548 |
T P |
| 2a0x_A |
282 |
191 |
168 |
1.97 |
10.71 |
65.375 |
8.120 |
T P |
| 1cb0_A |
268 |
191 |
168 |
1.94 |
8.33 |
65.207 |
8.230 |
T P |
| 1xe3_D |
235 |
191 |
158 |
1.92 |
12.66 |
64.511 |
7.831 |
T P |
| 3bgs_A |
286 |
191 |
167 |
2.00 |
10.78 |
64.303 |
7.940 |
T P |
| 1ybf_A |
240 |
191 |
145 |
1.95 |
18.62 |
63.904 |
7.089 |
T P |
| 1ovg_A |
237 |
191 |
154 |
1.83 |
9.74 |
63.855 |
7.994 |
T P |
| 2a8y_A |
263 |
191 |
164 |
1.96 |
12.20 |
63.504 |
7.973 |
T P |
| 2a0w_A |
282 |
191 |
165 |
1.99 |
10.30 |
63.373 |
7.896 |
T P |
| 2qpl_A |
266 |
191 |
155 |
2.00 |
10.97 |
63.100 |
7.396 |
T P |
| 1wta_A |
273 |
191 |
163 |
1.99 |
13.50 |
62.830 |
7.801 |
T P |
| 1v4n_B |
269 |
191 |
164 |
1.98 |
11.59 |
62.759 |
7.895 |
T P |
| 1je0_C |
227 |
191 |
157 |
2.07 |
12.10 |
62.430 |
7.247 |
T P |
| 2ac7_B |
221 |
191 |
150 |
1.90 |
12.67 |
62.303 |
7.519 |
T P |
| 2bsx_A |
241 |
191 |
149 |
1.81 |
10.74 |
62.301 |
7.785 |
T P |
| 2b94_A |
242 |
191 |
146 |
1.86 |
8.22 |
62.282 |
7.448 |
T P |
| 2ai2_A |
274 |
191 |
166 |
2.05 |
10.84 |
62.060 |
7.710 |
T P |
| 1z34_A |
230 |
191 |
155 |
2.01 |
11.61 |
62.000 |
7.344 |
T P |
| 1b8o_A |
280 |
191 |
164 |
2.01 |
9.76 |
61.862 |
7.760 |
T P |
| 1lvu_D |
279 |
191 |
159 |
2.01 |
10.06 |
61.700 |
7.518 |
T P |
| 2a0y_A |
282 |
191 |
163 |
1.96 |
10.43 |
61.683 |
7.927 |
T P |
| 1yqq_A |
273 |
191 |
164 |
2.02 |
9.76 |
61.482 |
7.753 |
T P |
| 1sq6_A |
225 |
191 |
145 |
1.84 |
10.34 |
61.274 |
7.482 |
T P |
| 1a9o_A |
289 |
191 |
164 |
2.05 |
10.37 |
60.955 |
7.617 |
T P |
| 1vfn_A |
270 |
191 |
161 |
2.01 |
11.18 |
60.859 |
7.640 |
T P |
| 1a9t_A |
284 |
191 |
162 |
2.06 |
9.88 |
60.819 |
7.489 |
T P |
| 1a9q_A |
282 |
191 |
162 |
2.03 |
10.49 |
60.777 |
7.613 |
T P |
| 1fxu_A |
278 |
191 |
162 |
2.07 |
10.49 |
60.405 |
7.454 |
T P |
| 1m73_E |
288 |
191 |
164 |
2.05 |
10.37 |
59.871 |
7.623 |
T P |
| 3pnp_A |
263 |
191 |
155 |
1.96 |
10.32 |
59.504 |
7.519 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]