LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_108.5wLII_11068_93
Total number of 3D structures: 50
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
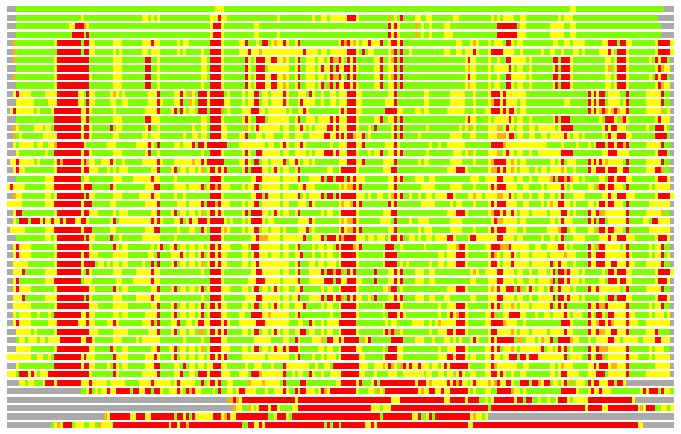
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1w8g_A |
226 |
227 |
221 |
0.49 |
24.89 |
96.862 |
37.556 |
T P |
| 3cpg_A |
253 |
227 |
214 |
1.53 |
24.77 |
87.797 |
13.146 |
T P |
| 1b54_A |
230 |
227 |
205 |
1.36 |
22.44 |
86.280 |
13.999 |
T P |
| 1ct5_A |
228 |
227 |
203 |
1.33 |
21.67 |
85.839 |
14.169 |
T P |
| 1vfs_A |
382 |
227 |
187 |
1.96 |
14.44 |
60.802 |
9.072 |
T P |
| 1xfc_A |
366 |
227 |
190 |
2.07 |
13.68 |
60.621 |
8.740 |
T P |
| 2rjg_A |
358 |
227 |
179 |
1.91 |
16.76 |
60.401 |
8.925 |
T P |
| 3b8w_A |
358 |
227 |
183 |
1.96 |
15.85 |
60.387 |
8.866 |
T P |
| 3b8t_A |
358 |
227 |
179 |
1.91 |
16.76 |
60.366 |
8.925 |
T P |
| 3b8v_A |
358 |
227 |
180 |
1.91 |
17.22 |
60.243 |
8.946 |
T P |
| 3c5q_A |
394 |
227 |
187 |
2.07 |
11.76 |
60.061 |
8.610 |
T P |
| 2qgh_A |
394 |
227 |
188 |
2.13 |
11.70 |
59.679 |
8.440 |
T P |
| 2p3e_B |
403 |
227 |
181 |
2.15 |
15.47 |
58.927 |
8.047 |
T P |
| 3b8u_A |
358 |
227 |
183 |
2.00 |
15.30 |
58.874 |
8.707 |
T P |
| 2odo_A |
354 |
227 |
185 |
2.05 |
12.97 |
58.228 |
8.592 |
T P |
| 2dy3_D |
344 |
227 |
189 |
2.18 |
13.23 |
57.639 |
8.279 |
T P |
| 2j66_A |
390 |
227 |
183 |
2.30 |
14.21 |
57.559 |
7.626 |
T P |
| 1rcq_A |
356 |
227 |
183 |
2.04 |
13.66 |
56.818 |
8.534 |
T P |
| 1tuf_A |
433 |
227 |
187 |
2.24 |
11.23 |
56.625 |
7.982 |
T P |
| 2nva_D |
371 |
227 |
177 |
2.20 |
14.69 |
56.124 |
7.701 |
T P |
| 1bd0_A |
381 |
227 |
182 |
2.14 |
16.48 |
55.823 |
8.116 |
T P |
| 2o0t_A |
445 |
227 |
190 |
2.21 |
15.26 |
55.791 |
8.219 |
T P |
| 2vd9_A |
386 |
227 |
183 |
2.20 |
15.30 |
55.774 |
7.958 |
T P |
| 1hkw_A |
446 |
227 |
190 |
2.19 |
15.79 |
55.660 |
8.313 |
T P |
| 2nv9_B |
372 |
227 |
181 |
2.23 |
13.26 |
55.632 |
7.763 |
T P |
| 1twi_A |
434 |
227 |
181 |
2.27 |
12.15 |
55.538 |
7.652 |
T P |
| 1hkv_A |
446 |
227 |
188 |
2.18 |
15.43 |
55.378 |
8.230 |
T P |
| 2vd8_A |
386 |
227 |
180 |
2.24 |
14.44 |
54.879 |
7.700 |
T P |
| 2plj_A |
376 |
227 |
180 |
2.24 |
13.33 |
54.771 |
7.694 |
T P |
| 1njj_D |
354 |
227 |
184 |
2.33 |
14.67 |
54.421 |
7.572 |
T P |
| 2on3_A |
392 |
227 |
179 |
2.32 |
14.53 |
54.382 |
7.404 |
T P |
| 1epv_A |
381 |
227 |
182 |
2.18 |
17.03 |
54.372 |
7.998 |
T P |
| 2sfp_A |
378 |
227 |
180 |
2.15 |
17.22 |
54.173 |
8.003 |
T P |
| 2oo0_A |
419 |
227 |
178 |
2.28 |
14.04 |
53.856 |
7.478 |
T P |
| 1xql_A |
381 |
227 |
181 |
2.18 |
17.13 |
53.828 |
7.923 |
T P |
| 7odc_A |
387 |
227 |
180 |
2.28 |
16.11 |
53.745 |
7.575 |
T P |
| 3btn_A |
394 |
227 |
178 |
2.23 |
13.48 |
53.468 |
7.649 |
T P |
| 2yxx_A |
385 |
227 |
179 |
2.33 |
16.76 |
53.189 |
7.351 |
T P |
| 2tod_A |
353 |
227 |
180 |
2.37 |
13.89 |
52.879 |
7.283 |
T P |
| 3co8_A |
360 |
227 |
179 |
2.33 |
13.97 |
52.183 |
7.379 |
T P |
| 1d7k_A |
411 |
227 |
179 |
2.31 |
14.53 |
52.113 |
7.422 |
T P |
| 1szr_D |
368 |
227 |
179 |
2.38 |
14.53 |
51.543 |
7.218 |
T P |
| 1f3t_B |
381 |
227 |
178 |
2.41 |
14.61 |
51.206 |
7.105 |
T P |
| 1knw_A |
421 |
227 |
177 |
2.38 |
15.25 |
49.728 |
7.137 |
T P |
| 1lt8_A |
348 |
227 |
142 |
2.55 |
5.63 |
37.380 |
5.366 |
T P |
| 2c6q_B |
329 |
227 |
132 |
2.41 |
8.33 |
37.360 |
5.259 |
T P |
| 1py0_A |
122 |
227 |
35 |
2.54 |
8.57 |
10.766 |
1.325 |
T P |
| 2g98_A |
171 |
227 |
37 |
2.69 |
10.81 |
10.544 |
1.325 |
T P |
| 1h4a_X |
173 |
227 |
37 |
2.82 |
13.51 |
10.357 |
1.267 |
T P |
| 1hk0_X |
173 |
227 |
36 |
2.89 |
8.33 |
10.045 |
1.205 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]