LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_109.5wLII_11068_97
Total number of 3D structures: 37
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
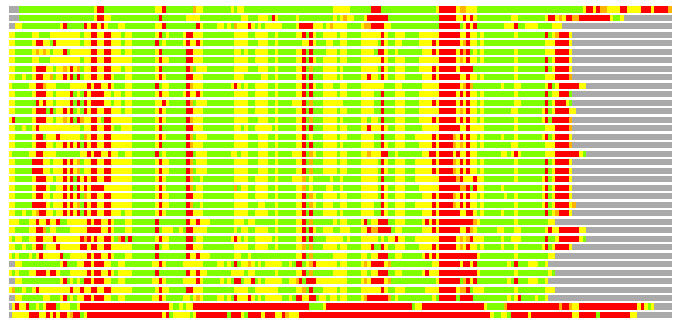
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ihm_B |
511 |
194 |
168 |
1.66 |
18.45 |
81.945 |
9.570 |
T P |
| 2gh8_A |
544 |
194 |
147 |
1.81 |
17.01 |
67.928 |
7.696 |
T P |
| 3cji_C |
268 |
194 |
135 |
2.08 |
8.15 |
50.852 |
6.182 |
T P |
| 1pvc_3 |
235 |
194 |
144 |
2.19 |
10.42 |
49.995 |
6.290 |
T P |
| 1pov_3 |
238 |
194 |
141 |
2.12 |
8.51 |
49.930 |
6.358 |
T P |
| 1fpn_3 |
237 |
194 |
141 |
2.13 |
9.22 |
49.736 |
6.309 |
T P |
| 1vbb_3 |
235 |
194 |
144 |
2.21 |
11.11 |
49.720 |
6.233 |
T P |
| 1cov_3 |
238 |
194 |
138 |
2.13 |
13.04 |
49.445 |
6.191 |
T P |
| 1h8t_C |
238 |
194 |
137 |
2.12 |
11.68 |
49.322 |
6.182 |
T P |
| 1mec_3 |
231 |
194 |
144 |
2.22 |
6.94 |
49.319 |
6.202 |
T P |
| 2rhn_3 |
238 |
194 |
139 |
2.16 |
7.91 |
49.303 |
6.155 |
T P |
| 1r1a_3 |
238 |
194 |
140 |
2.17 |
7.86 |
49.181 |
6.164 |
T P |
| 1aym_3 |
238 |
194 |
138 |
2.14 |
9.42 |
49.160 |
6.174 |
T P |
| 1bev_3 |
242 |
194 |
137 |
2.12 |
9.49 |
49.110 |
6.184 |
T P |
| 1d4m_3 |
238 |
194 |
142 |
2.19 |
11.27 |
49.062 |
6.191 |
T P |
| 1z7s_3 |
239 |
194 |
142 |
2.21 |
11.27 |
49.002 |
6.148 |
T P |
| 1oop_C |
238 |
194 |
139 |
2.15 |
12.23 |
48.913 |
6.190 |
T P |
| 1tme_3 |
230 |
194 |
141 |
2.22 |
9.22 |
48.636 |
6.077 |
T P |
| 3epf_3 |
235 |
194 |
140 |
2.22 |
10.00 |
48.509 |
6.043 |
T P |
| 1eah_3 |
235 |
194 |
140 |
2.22 |
10.00 |
48.509 |
6.043 |
T P |
| 1rhi_3 |
236 |
194 |
139 |
2.22 |
10.79 |
48.351 |
5.993 |
T P |
| 1ncq_C |
236 |
194 |
138 |
2.23 |
9.42 |
48.278 |
5.934 |
T P |
| 1hxs_3 |
235 |
194 |
139 |
2.19 |
10.07 |
48.210 |
6.071 |
T P |
| 3epc_3 |
235 |
194 |
139 |
2.21 |
10.07 |
48.157 |
6.010 |
T P |
| 1mqt_C |
238 |
194 |
136 |
2.10 |
11.76 |
48.130 |
6.188 |
T P |
| 1fhp_3 |
220 |
194 |
133 |
2.08 |
8.27 |
47.933 |
6.104 |
T P |
| 2mev_3 |
231 |
194 |
137 |
2.17 |
8.03 |
47.825 |
6.022 |
T P |
| 1tmf_3 |
232 |
194 |
139 |
2.24 |
7.91 |
47.736 |
5.951 |
T P |
| 1z7z_3 |
192 |
194 |
139 |
2.21 |
11.51 |
47.623 |
6.009 |
T P |
| 1zba_3 |
221 |
194 |
134 |
2.09 |
9.70 |
47.612 |
6.109 |
T P |
| 1zba_2 |
207 |
194 |
131 |
2.14 |
9.16 |
47.098 |
5.860 |
T P |
| 1fmd_3 |
219 |
194 |
132 |
2.08 |
8.33 |
47.053 |
6.045 |
T P |
| 1fhp_2 |
216 |
194 |
128 |
2.08 |
8.59 |
47.014 |
5.878 |
T P |
| 1ev1_3 |
239 |
194 |
142 |
2.32 |
11.97 |
46.859 |
5.879 |
T P |
| 1fmd_2 |
218 |
194 |
127 |
2.12 |
9.45 |
45.096 |
5.718 |
T P |
| 3by1_A |
280 |
194 |
45 |
2.53 |
6.67 |
16.792 |
1.713 |
T P |
| 3by2_A |
280 |
194 |
44 |
2.85 |
9.09 |
14.563 |
1.492 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]