LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_113.5wLII_11068_119
Total number of 3D structures: 23
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
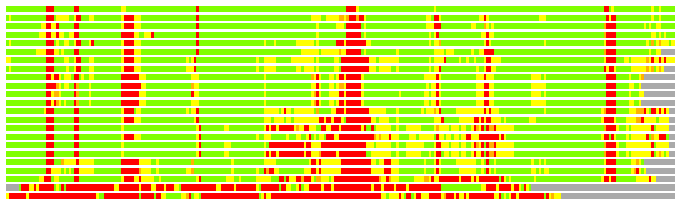
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2h12_B |
426 |
267 |
255 |
0.69 |
25.10 |
94.177 |
32.228 |
T P |
| 2r26_A |
381 |
267 |
246 |
1.42 |
21.54 |
87.010 |
16.193 |
T P |
| 1aj8_A |
371 |
267 |
244 |
1.30 |
25.00 |
86.522 |
17.448 |
T P |
| 2p2w_A |
358 |
267 |
243 |
1.30 |
20.99 |
86.339 |
17.367 |
T P |
| 2ifc_C |
382 |
267 |
240 |
1.50 |
21.67 |
83.654 |
14.957 |
T P |
| 1a59_A |
377 |
267 |
238 |
1.59 |
21.01 |
82.121 |
14.090 |
T P |
| 1vgm_A |
376 |
267 |
236 |
1.76 |
24.58 |
80.805 |
12.702 |
T P |
| 1o7x_C |
370 |
267 |
236 |
1.76 |
23.73 |
80.625 |
12.679 |
T P |
| 1csh_A |
435 |
267 |
227 |
1.54 |
16.74 |
80.508 |
13.875 |
T P |
| 1al6_A |
435 |
267 |
227 |
1.56 |
17.18 |
80.411 |
13.707 |
T P |
| 2cts_A |
437 |
267 |
228 |
1.58 |
16.67 |
80.372 |
13.554 |
T P |
| 1csc_A |
429 |
267 |
227 |
1.57 |
17.18 |
80.335 |
13.609 |
T P |
| 1vgp_A |
373 |
267 |
230 |
1.87 |
22.17 |
77.439 |
11.691 |
T P |
| 2ibp_A |
407 |
267 |
227 |
1.88 |
27.31 |
76.365 |
11.463 |
T P |
| 1owb_A |
426 |
267 |
224 |
1.83 |
21.88 |
76.347 |
11.597 |
T P |
| 1iom_A |
374 |
267 |
217 |
1.87 |
26.27 |
73.165 |
11.001 |
T P |
| 1k3p_A |
426 |
267 |
214 |
1.80 |
21.96 |
73.121 |
11.271 |
T P |
| 1nxe_A |
426 |
267 |
216 |
1.84 |
21.76 |
64.295 |
11.136 |
T P |
| 3enj_A |
437 |
267 |
220 |
1.90 |
17.73 |
63.063 |
11.013 |
T P |
| 5csc_B |
429 |
267 |
217 |
1.89 |
17.05 |
62.613 |
10.927 |
T P |
| 2c6x_A |
363 |
267 |
194 |
1.95 |
22.68 |
55.602 |
9.467 |
T P |
| 1wcg_A |
462 |
267 |
61 |
2.46 |
1.64 |
15.655 |
2.382 |
T P |
| 2pul_A |
381 |
267 |
59 |
2.72 |
5.08 |
14.054 |
2.092 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]