LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_116.5wLII_11077_6
Total number of 3D structures: 96
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
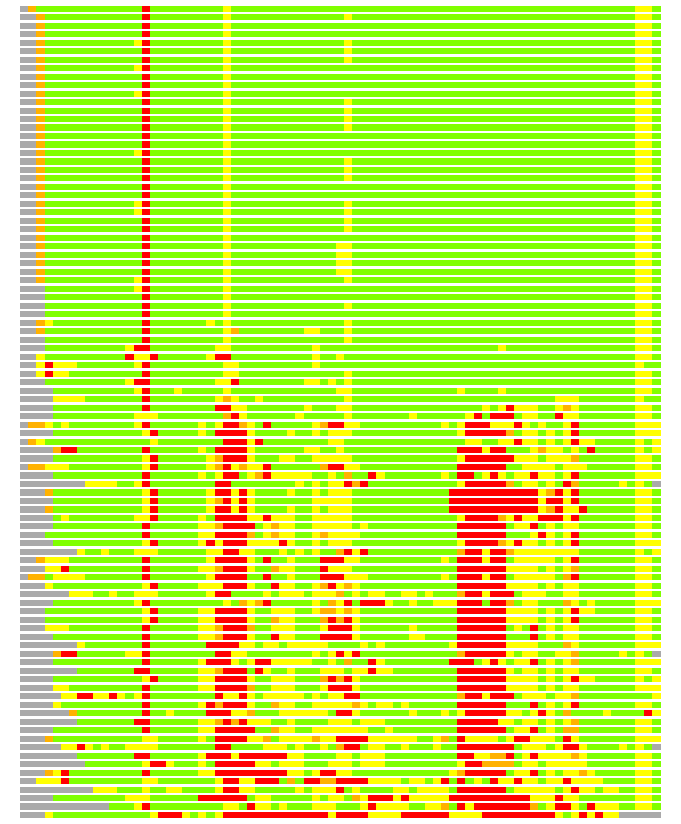
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ccr_A |
111 |
79 |
77 |
1.10 |
25.97 |
94.742 |
6.443 |
T P |
| 1ctz_A |
108 |
79 |
76 |
0.98 |
19.74 |
94.692 |
7.047 |
T P |
| 1cri_A |
108 |
79 |
76 |
0.97 |
18.42 |
94.409 |
7.078 |
T P |
| 2ycc_A |
108 |
79 |
76 |
1.00 |
19.74 |
94.404 |
6.917 |
T P |
| 1crh_A |
108 |
79 |
76 |
0.99 |
19.74 |
94.326 |
6.963 |
T P |
| 1chh_A |
108 |
79 |
76 |
0.99 |
19.74 |
94.326 |
6.995 |
T P |
| 1crg_A |
108 |
79 |
76 |
0.97 |
19.74 |
94.326 |
7.123 |
T P |
| 1csw_A |
108 |
79 |
76 |
1.00 |
19.74 |
94.309 |
6.936 |
T P |
| 1irw_A |
108 |
79 |
76 |
1.00 |
19.74 |
94.309 |
6.911 |
T P |
| 1irv_A |
108 |
79 |
76 |
1.02 |
21.05 |
94.298 |
6.759 |
T P |
| 1cih_A |
108 |
79 |
76 |
1.00 |
18.42 |
94.248 |
6.940 |
T P |
| 2b12_B |
108 |
79 |
76 |
1.01 |
19.74 |
94.226 |
6.860 |
T P |
| 1cie_A |
108 |
79 |
76 |
0.99 |
19.74 |
94.226 |
6.947 |
T P |
| 1ycc_A |
108 |
79 |
76 |
0.99 |
19.74 |
94.226 |
6.956 |
T P |
| 2jqr_A |
108 |
79 |
76 |
0.99 |
19.74 |
94.226 |
6.956 |
T P |
| 1raq_A |
108 |
79 |
76 |
1.01 |
19.74 |
94.154 |
6.876 |
T P |
| 1cig_A |
108 |
79 |
76 |
0.99 |
18.42 |
94.154 |
6.962 |
T P |
| 1csu_A |
108 |
79 |
76 |
1.00 |
19.74 |
94.126 |
6.892 |
T P |
| 1rap_A |
108 |
79 |
76 |
0.98 |
19.74 |
94.126 |
7.033 |
T P |
| 2b11_B |
108 |
79 |
76 |
1.03 |
19.74 |
94.121 |
6.736 |
T P |
| 2bcn_B |
108 |
79 |
76 |
1.05 |
19.74 |
94.115 |
6.607 |
T P |
| 1cif_A |
108 |
79 |
76 |
1.00 |
19.74 |
94.115 |
6.882 |
T P |
| 1csx_A |
108 |
79 |
76 |
1.02 |
22.37 |
94.054 |
6.791 |
T P |
| 1chi_A |
108 |
79 |
76 |
1.02 |
21.05 |
94.054 |
6.789 |
T P |
| 1chj_A |
108 |
79 |
76 |
1.02 |
21.05 |
94.026 |
6.756 |
T P |
| 1s6v_B |
108 |
79 |
76 |
1.07 |
21.05 |
93.965 |
6.485 |
T P |
| 2b0z_B |
108 |
79 |
76 |
1.03 |
19.74 |
93.937 |
6.702 |
T P |
| 1csv_A |
108 |
79 |
76 |
1.02 |
19.74 |
93.926 |
6.801 |
T P |
| 1ytc_A |
112 |
79 |
76 |
1.08 |
18.42 |
93.843 |
6.448 |
T P |
| 1yeb_A |
108 |
79 |
76 |
1.03 |
21.05 |
93.843 |
6.753 |
T P |
| 3cxh_W |
112 |
79 |
76 |
1.11 |
18.42 |
93.771 |
6.295 |
T P |
| 1yea_A |
112 |
79 |
76 |
1.07 |
19.74 |
93.743 |
6.519 |
T P |
| 3cx5_W |
108 |
79 |
76 |
1.09 |
19.74 |
93.676 |
6.402 |
T P |
| 2aiu_A |
104 |
79 |
75 |
0.78 |
29.33 |
93.510 |
8.567 |
T P |
| 1i54_A |
103 |
79 |
75 |
0.79 |
24.00 |
93.415 |
8.425 |
T P |
| 2b4z_A |
104 |
79 |
75 |
0.84 |
26.67 |
93.316 |
7.945 |
T P |
| 1wej_F |
104 |
79 |
75 |
0.83 |
26.67 |
93.316 |
8.051 |
T P |
| 2b10_B |
108 |
79 |
76 |
1.19 |
19.74 |
92.816 |
5.911 |
T P |
| 1hro_A |
105 |
79 |
76 |
1.34 |
22.37 |
92.155 |
5.295 |
T P |
| 2jti_B |
103 |
79 |
75 |
1.09 |
20.00 |
91.411 |
6.314 |
T P |
| 1co6_A |
107 |
79 |
74 |
1.05 |
24.32 |
90.778 |
6.413 |
T P |
| 1qn2_A |
99 |
79 |
73 |
1.05 |
26.03 |
89.435 |
6.363 |
T P |
| 1yfc_A |
108 |
79 |
75 |
1.44 |
21.33 |
89.229 |
4.873 |
T P |
| 1fhb_A |
108 |
79 |
75 |
1.35 |
20.00 |
89.057 |
5.171 |
T P |
| 1ql3_A |
99 |
79 |
73 |
1.18 |
28.77 |
88.757 |
5.719 |
T P |
| 1cyc_A |
103 |
79 |
74 |
1.39 |
24.32 |
86.998 |
4.975 |
T P |
| 1j3s_A |
104 |
79 |
74 |
1.66 |
25.68 |
85.704 |
4.213 |
T P |
| 3cp5_A |
116 |
79 |
71 |
1.72 |
19.72 |
82.073 |
3.896 |
T P |
| 1w2l_A |
97 |
79 |
69 |
1.67 |
20.29 |
81.393 |
3.896 |
T P |
| 1umm_A |
149 |
79 |
67 |
2.06 |
14.93 |
75.024 |
3.103 |
T P |
| 1hzu_A |
521 |
79 |
64 |
1.84 |
12.50 |
73.425 |
3.306 |
T P |
| 2c1d_B |
137 |
79 |
72 |
1.81 |
22.22 |
73.046 |
3.764 |
T P |
| 1b7c_A |
71 |
79 |
62 |
1.85 |
22.58 |
71.008 |
3.181 |
T P |
| 1e2r_B |
543 |
79 |
65 |
1.95 |
13.85 |
66.123 |
3.166 |
T P |
| 2d0w_A |
168 |
79 |
67 |
2.21 |
17.91 |
65.327 |
2.897 |
T P |
| 1kv9_A |
664 |
79 |
65 |
1.98 |
20.00 |
64.483 |
3.125 |
T P |
| 2d0s_A |
79 |
79 |
59 |
1.98 |
15.25 |
64.154 |
2.840 |
T P |
| 1gq1_A |
559 |
79 |
59 |
1.94 |
13.56 |
62.913 |
2.895 |
T P |
| 1hj5_A |
559 |
79 |
58 |
1.79 |
13.79 |
62.579 |
3.066 |
T P |
| 1qks_A |
559 |
79 |
59 |
1.99 |
13.56 |
62.508 |
2.822 |
T P |
| 1hzv_A |
514 |
79 |
60 |
2.00 |
13.33 |
61.818 |
2.863 |
T P |
| 2v08_A |
84 |
79 |
63 |
2.03 |
14.29 |
61.560 |
2.964 |
T P |
| 2zbo_A |
86 |
79 |
63 |
2.04 |
14.29 |
61.353 |
2.950 |
T P |
| 1nir_B |
539 |
79 |
63 |
2.03 |
12.70 |
60.713 |
2.953 |
T P |
| 2i8f_A |
82 |
79 |
64 |
2.17 |
14.06 |
60.503 |
2.817 |
T P |
| 2gc4_D |
147 |
79 |
62 |
1.96 |
17.74 |
60.407 |
3.016 |
T P |
| 1gdv_A |
85 |
79 |
64 |
2.16 |
15.62 |
60.389 |
2.827 |
T P |
| 1mg2_D |
147 |
79 |
63 |
2.12 |
15.87 |
60.296 |
2.842 |
T P |
| 1ctj_A |
89 |
79 |
64 |
2.19 |
17.19 |
60.255 |
2.795 |
T P |
| 1fi3_A |
82 |
79 |
66 |
2.18 |
13.64 |
60.201 |
2.889 |
T P |
| 1kb0_A |
669 |
79 |
64 |
2.08 |
18.75 |
59.822 |
2.941 |
T P |
| 1cyj_A |
90 |
79 |
64 |
2.21 |
17.19 |
59.134 |
2.774 |
T P |
| 1c6r_A |
88 |
79 |
62 |
2.11 |
14.52 |
59.067 |
2.806 |
T P |
| 2ce0_A |
99 |
79 |
62 |
2.12 |
17.74 |
59.024 |
2.790 |
T P |
| 2dge_A |
102 |
79 |
59 |
1.93 |
18.64 |
58.971 |
2.904 |
T P |
| 1kx2_A |
81 |
79 |
61 |
2.16 |
16.39 |
58.949 |
2.704 |
T P |
| 351c_A |
82 |
79 |
62 |
1.98 |
16.13 |
58.810 |
2.975 |
T P |
| 1yiq_A |
684 |
79 |
63 |
1.98 |
20.63 |
58.396 |
3.028 |
T P |
| 1c52_A |
131 |
79 |
59 |
1.97 |
25.42 |
58.230 |
2.844 |
T P |
| 1ls9_A |
91 |
79 |
61 |
2.10 |
19.67 |
58.190 |
2.770 |
T P |
| 2v07_A |
98 |
79 |
61 |
2.07 |
16.39 |
58.120 |
2.813 |
T P |
| 1cch_A |
82 |
79 |
61 |
2.17 |
13.11 |
57.949 |
2.685 |
T P |
| 1kib_A |
89 |
79 |
62 |
2.23 |
14.52 |
57.938 |
2.664 |
T P |
| 1wve_C |
75 |
79 |
60 |
2.21 |
21.67 |
57.884 |
2.602 |
T P |
| 1dt1_A |
129 |
79 |
63 |
2.15 |
25.40 |
56.629 |
2.800 |
T P |
| 1f1f_A |
88 |
79 |
62 |
2.09 |
14.52 |
56.457 |
2.829 |
T P |
| 1m70_A |
190 |
79 |
64 |
2.35 |
14.06 |
55.184 |
2.615 |
T P |
| 1cor_A |
82 |
79 |
59 |
2.07 |
15.25 |
55.104 |
2.720 |
T P |
| 2fwl_A |
129 |
79 |
57 |
2.19 |
26.32 |
54.986 |
2.493 |
T P |
| 1foc_A |
128 |
79 |
62 |
2.28 |
24.19 |
53.414 |
2.604 |
T P |
| 1c6s_A |
87 |
79 |
57 |
2.12 |
15.79 |
52.381 |
2.563 |
T P |
| 1h1o_A |
172 |
79 |
60 |
2.39 |
8.33 |
52.034 |
2.406 |
T P |
| 1a56_A |
81 |
79 |
60 |
2.32 |
11.67 |
51.620 |
2.483 |
T P |
| 2c1v_A |
335 |
79 |
54 |
2.13 |
9.26 |
49.976 |
2.426 |
T P |
| 1lms_A |
95 |
79 |
54 |
2.27 |
18.52 |
48.290 |
2.280 |
T P |
| 1gpe_A |
587 |
79 |
34 |
2.45 |
2.94 |
32.229 |
1.333 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]