LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_12.5wLII_09933_2
Total number of 3D structures: 48
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
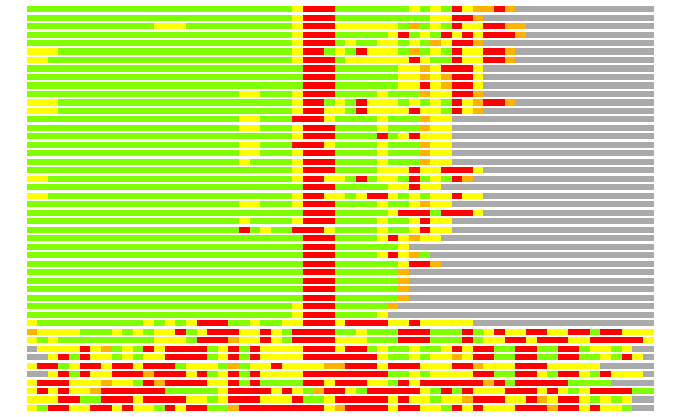
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3com_B |
281 |
59 |
41 |
1.91 |
21.95 |
65.766 |
2.040 |
T P |
| 2j7t_A |
285 |
59 |
38 |
1.28 |
42.11 |
62.682 |
2.747 |
T P |
| 2j0i_A |
290 |
59 |
41 |
2.31 |
26.83 |
62.098 |
1.700 |
T P |
| 2ja0_A |
286 |
59 |
38 |
1.76 |
26.32 |
62.080 |
2.045 |
T P |
| 3ckw_A |
255 |
59 |
38 |
1.69 |
21.05 |
60.824 |
2.127 |
T P |
| 2bva_B |
275 |
59 |
40 |
2.14 |
22.50 |
60.600 |
1.785 |
T P |
| 1yhv_A |
293 |
59 |
39 |
2.04 |
17.95 |
60.345 |
1.819 |
T P |
| 1s9j_A |
289 |
59 |
37 |
1.63 |
13.51 |
60.326 |
2.141 |
T P |
| 3eqc_A |
312 |
59 |
38 |
1.91 |
13.16 |
60.134 |
1.886 |
T P |
| 3eqb_A |
289 |
59 |
37 |
1.72 |
13.51 |
60.134 |
2.030 |
T P |
| 2f7x_E |
336 |
59 |
38 |
1.82 |
10.53 |
59.686 |
1.975 |
T P |
| 2c30_A |
290 |
59 |
40 |
2.23 |
17.50 |
59.396 |
1.718 |
T P |
| 1yhw_A |
293 |
59 |
38 |
1.89 |
18.42 |
59.326 |
1.914 |
T P |
| 1apm_E |
341 |
59 |
37 |
1.67 |
10.81 |
59.134 |
2.094 |
T P |
| 1cdk_A |
343 |
59 |
37 |
1.71 |
10.81 |
59.114 |
2.047 |
T P |
| 2f57_B |
300 |
59 |
35 |
1.43 |
25.71 |
59.077 |
2.285 |
T P |
| 1ydr_E |
336 |
59 |
37 |
1.68 |
10.81 |
59.001 |
2.074 |
T P |
| 1atp_E |
336 |
59 |
37 |
1.71 |
10.81 |
58.995 |
2.049 |
T P |
| 2c1a_A |
337 |
59 |
37 |
1.70 |
10.81 |
58.861 |
2.054 |
T P |
| 2q0n_A |
294 |
59 |
36 |
1.62 |
30.56 |
58.861 |
2.095 |
T P |
| 3fxz_A |
293 |
59 |
37 |
1.76 |
18.92 |
58.646 |
1.992 |
T P |
| 1u5q_A |
309 |
59 |
35 |
1.39 |
14.29 |
58.608 |
2.343 |
T P |
| 3ckx_A |
277 |
59 |
38 |
1.86 |
18.42 |
58.358 |
1.935 |
T P |
| 2qur_A |
340 |
59 |
37 |
1.80 |
10.81 |
57.672 |
1.945 |
T P |
| 2gcd_A |
309 |
59 |
34 |
1.10 |
14.71 |
57.441 |
2.825 |
T P |
| 2erz_E |
336 |
59 |
36 |
1.51 |
11.11 |
57.040 |
2.241 |
T P |
| 1syk_A |
350 |
59 |
35 |
1.41 |
11.43 |
57.034 |
2.316 |
T P |
| 2vwi_D |
284 |
59 |
35 |
1.55 |
17.14 |
56.638 |
2.127 |
T P |
| 2qr8_A |
298 |
59 |
33 |
1.02 |
21.21 |
55.538 |
2.939 |
T P |
| 3dak_D |
284 |
59 |
34 |
1.47 |
17.65 |
55.508 |
2.161 |
T P |
| 1kob_A |
352 |
59 |
34 |
1.66 |
17.65 |
54.847 |
1.933 |
T P |
| 2jc6_A |
278 |
59 |
33 |
1.34 |
9.09 |
54.579 |
2.290 |
T P |
| 2jam_B |
280 |
59 |
33 |
1.41 |
15.15 |
54.319 |
2.191 |
T P |
| 2hw7_A |
271 |
59 |
33 |
1.19 |
27.27 |
54.148 |
2.553 |
T P |
| 1a06_A |
279 |
59 |
33 |
1.37 |
9.09 |
54.051 |
2.243 |
T P |
| 2hw6_A |
242 |
59 |
32 |
1.25 |
28.12 |
52.751 |
2.366 |
T P |
| 2ac3_A |
277 |
59 |
31 |
0.99 |
29.03 |
51.234 |
2.850 |
T P |
| 2qr7_A |
298 |
59 |
31 |
2.18 |
6.45 |
45.403 |
1.360 |
T P |
| 2j51_A |
288 |
59 |
39 |
2.39 |
10.26 |
44.978 |
1.564 |
T P |
| 2jfl_A |
288 |
59 |
37 |
2.33 |
8.11 |
44.135 |
1.524 |
T P |
| 1dar_A |
615 |
59 |
36 |
2.59 |
2.78 |
40.185 |
1.338 |
T P |
| 2efg_A |
582 |
59 |
34 |
2.55 |
5.88 |
38.894 |
1.284 |
T P |
| 2h5e_A |
488 |
59 |
33 |
3.01 |
9.09 |
37.803 |
1.061 |
T P |
| 2j7k_A |
660 |
59 |
30 |
2.54 |
3.33 |
36.147 |
1.138 |
T P |
| 2bm0_A |
666 |
59 |
27 |
2.63 |
3.70 |
34.711 |
0.989 |
T P |
| 2bm1_A |
660 |
59 |
29 |
2.56 |
3.45 |
33.346 |
1.089 |
T P |
| 2bv3_A |
632 |
59 |
32 |
2.87 |
0.00 |
32.702 |
1.077 |
T P |
| 1fnm_A |
655 |
59 |
27 |
2.82 |
11.11 |
31.566 |
0.926 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]