LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_120.5wLII_11077_13
Total number of 3D structures: 67
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
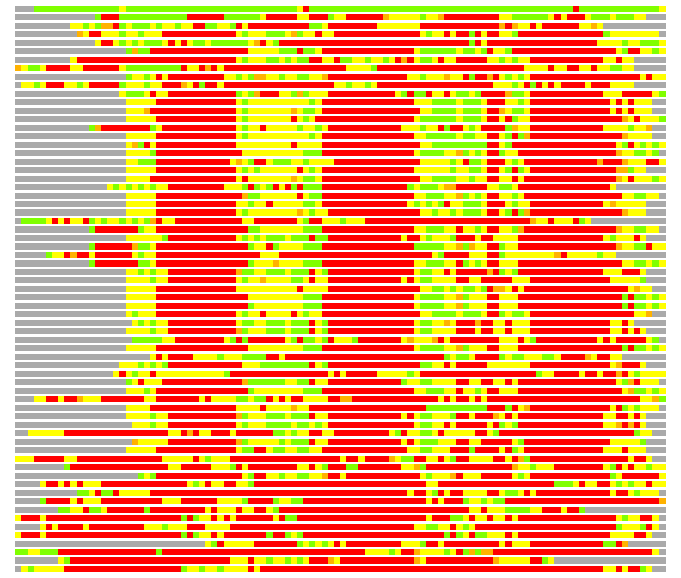
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2r39_A |
109 |
106 |
101 |
0.55 |
17.82 |
94.704 |
15.433 |
T P |
| 1ti6_B |
274 |
106 |
54 |
2.22 |
5.56 |
37.092 |
2.325 |
T P |
| 3c7b_A |
417 |
106 |
44 |
2.68 |
4.55 |
29.494 |
1.584 |
T P |
| 2vkr_A |
103 |
106 |
46 |
2.92 |
10.87 |
26.653 |
1.522 |
T P |
| 2c42_A |
1231 |
106 |
38 |
2.35 |
7.89 |
26.363 |
1.554 |
T P |
| 1b0v_A |
106 |
106 |
36 |
2.22 |
2.78 |
26.218 |
1.555 |
T P |
| 2fug_9 |
154 |
106 |
42 |
2.66 |
7.14 |
25.917 |
1.524 |
T P |
| 2vpz_B |
193 |
106 |
40 |
2.66 |
12.50 |
25.860 |
1.448 |
T P |
| 1jnr_B |
149 |
106 |
42 |
2.83 |
4.76 |
25.480 |
1.435 |
T P |
| 1kqf_B |
289 |
106 |
39 |
2.86 |
2.56 |
25.213 |
1.318 |
T P |
| 1blu_A |
80 |
106 |
39 |
2.64 |
7.69 |
24.884 |
1.425 |
T P |
| 1gao_A |
106 |
106 |
40 |
2.73 |
0.00 |
24.757 |
1.414 |
T P |
| 1dur_A |
55 |
106 |
39 |
2.51 |
2.56 |
24.727 |
1.495 |
T P |
| 2fgo_A |
81 |
106 |
39 |
2.56 |
2.56 |
24.593 |
1.468 |
T P |
| 1f5c_A |
106 |
106 |
40 |
2.73 |
12.50 |
24.537 |
1.416 |
T P |
| 7fd1_A |
106 |
106 |
40 |
2.79 |
2.50 |
24.443 |
1.384 |
T P |
| 1bc6_A |
77 |
106 |
37 |
2.44 |
0.00 |
24.424 |
1.454 |
T P |
| 1d3w_A |
106 |
106 |
40 |
2.65 |
10.00 |
24.384 |
1.456 |
T P |
| 1jb0_C |
80 |
106 |
39 |
2.80 |
7.69 |
24.199 |
1.344 |
T P |
| 1frj_A |
106 |
106 |
39 |
2.80 |
2.56 |
24.177 |
1.345 |
T P |
| 1bqx_A |
77 |
106 |
38 |
2.72 |
0.00 |
24.169 |
1.346 |
T P |
| 3c7b_B |
363 |
106 |
36 |
2.41 |
2.78 |
24.102 |
1.436 |
T P |
| 1b0t_A |
106 |
106 |
39 |
2.68 |
5.13 |
24.063 |
1.404 |
T P |
| 2fdn_A |
55 |
106 |
41 |
2.78 |
14.63 |
23.984 |
1.422 |
T P |
| 1frh_A |
106 |
106 |
39 |
2.79 |
2.56 |
23.888 |
1.349 |
T P |
| 3bk7_A |
593 |
106 |
40 |
2.64 |
7.50 |
23.880 |
1.458 |
T P |
| 1f5b_A |
106 |
106 |
38 |
2.58 |
5.26 |
23.844 |
1.419 |
T P |
| 1h98_A |
77 |
106 |
39 |
2.75 |
7.69 |
23.791 |
1.368 |
T P |
| 1fdd_A |
106 |
106 |
38 |
2.57 |
5.26 |
23.728 |
1.423 |
T P |
| 2ivf_B |
337 |
106 |
35 |
2.95 |
5.71 |
23.721 |
1.147 |
T P |
| 1a6l_A |
106 |
106 |
37 |
2.52 |
2.70 |
23.679 |
1.412 |
T P |
| 1ftc_A |
106 |
106 |
35 |
2.62 |
5.71 |
23.625 |
1.289 |
T P |
| 1fri_A |
106 |
106 |
40 |
2.81 |
10.00 |
23.619 |
1.376 |
T P |
| 2fd2_A |
106 |
106 |
38 |
2.81 |
0.00 |
23.503 |
1.304 |
T P |
| 1frl_A |
106 |
106 |
38 |
2.53 |
7.89 |
23.498 |
1.445 |
T P |
| 1frm_A |
106 |
106 |
38 |
2.53 |
7.89 |
23.472 |
1.446 |
T P |
| 1fca_A |
55 |
106 |
36 |
2.42 |
5.56 |
23.472 |
1.431 |
T P |
| 1pc4_A |
106 |
106 |
36 |
2.81 |
5.56 |
23.463 |
1.238 |
T P |
| 1pc5_A |
106 |
106 |
38 |
2.84 |
5.26 |
23.448 |
1.292 |
T P |
| 1rgv_A |
80 |
106 |
36 |
2.54 |
5.56 |
23.438 |
1.364 |
T P |
| 1frk_A |
106 |
106 |
38 |
2.53 |
7.89 |
23.351 |
1.443 |
T P |
| 2v4j_A |
436 |
106 |
34 |
2.50 |
5.88 |
23.210 |
1.306 |
T P |
| 1ff2_A |
106 |
106 |
36 |
2.72 |
5.56 |
23.149 |
1.275 |
T P |
| 1hfe_L |
396 |
106 |
38 |
3.04 |
5.26 |
23.140 |
1.210 |
T P |
| 1fd2_A |
106 |
106 |
38 |
2.68 |
10.53 |
23.012 |
1.367 |
T P |
| 1g3o_A |
106 |
106 |
38 |
2.60 |
10.53 |
22.973 |
1.407 |
T P |
| 2c3m_A |
1231 |
106 |
35 |
3.02 |
14.29 |
22.932 |
1.122 |
T P |
| 1clf_A |
55 |
106 |
35 |
2.56 |
2.86 |
22.902 |
1.315 |
T P |
| 2v2k_A |
104 |
106 |
37 |
2.86 |
5.41 |
22.896 |
1.248 |
T P |
| 1frx_A |
106 |
106 |
36 |
2.77 |
5.56 |
22.869 |
1.256 |
T P |
| 1y4z_B |
509 |
106 |
37 |
2.84 |
5.41 |
22.492 |
1.257 |
T P |
| 1g6b_A |
106 |
106 |
38 |
2.81 |
7.89 |
22.358 |
1.304 |
T P |
| 1fdx |
54 |
106 |
34 |
2.66 |
8.82 |
22.221 |
1.231 |
T P |
| 3c8y_A |
574 |
106 |
34 |
2.95 |
2.94 |
20.669 |
1.115 |
T P |
| 2o01_C |
80 |
106 |
33 |
2.78 |
3.03 |
20.537 |
1.146 |
T P |
| 1k0t_A |
80 |
106 |
31 |
2.88 |
6.45 |
20.531 |
1.039 |
T P |
| 1yy9_A |
613 |
106 |
30 |
2.77 |
6.67 |
19.375 |
1.046 |
T P |
| 1xer_A |
102 |
106 |
29 |
2.90 |
3.45 |
18.383 |
0.968 |
T P |
| 3b2v_A |
375 |
106 |
28 |
2.66 |
10.71 |
18.262 |
1.016 |
T P |
| 3cf4_A |
766 |
106 |
29 |
2.73 |
10.34 |
17.616 |
1.024 |
T P |
| 1h7x_B |
1019 |
106 |
25 |
2.88 |
4.00 |
17.341 |
0.838 |
T P |
| 1e08_A |
371 |
106 |
29 |
2.87 |
3.45 |
17.246 |
0.975 |
T P |
| 1gte_D |
1014 |
106 |
25 |
2.83 |
4.00 |
16.865 |
0.853 |
T P |
| 1h0h_B |
214 |
106 |
26 |
2.75 |
11.54 |
16.536 |
0.913 |
T P |
| 1igr_A |
471 |
106 |
27 |
2.83 |
0.00 |
16.367 |
0.921 |
T P |
| 1q16_B |
509 |
106 |
26 |
2.95 |
3.85 |
16.283 |
0.853 |
T P |
| 1nql_A |
612 |
106 |
24 |
2.86 |
0.00 |
14.393 |
0.811 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]