LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_124.5wLII_11077_30
Total number of 3D structures: 42
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
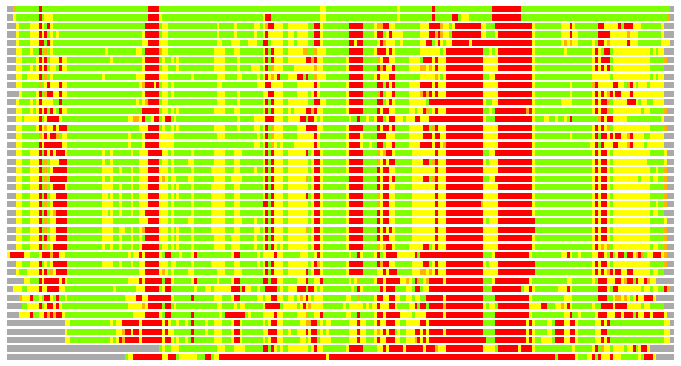
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2qvp_C |
269 |
232 |
213 |
0.57 |
29.11 |
91.130 |
31.962 |
T P |
| 3b2y_A |
262 |
232 |
209 |
0.92 |
25.36 |
88.086 |
20.440 |
T P |
| 2nsm_A |
390 |
232 |
175 |
1.95 |
25.14 |
59.913 |
8.520 |
T P |
| 1h8l_A |
380 |
232 |
178 |
2.03 |
20.79 |
58.523 |
8.372 |
T P |
| 1uwy_A |
393 |
232 |
179 |
2.12 |
24.58 |
58.493 |
8.055 |
T P |
| 1jqg_A |
409 |
232 |
181 |
2.07 |
17.68 |
57.780 |
8.354 |
T P |
| 1nsa_A |
395 |
232 |
174 |
2.09 |
17.24 |
56.107 |
7.944 |
T P |
| 1kwm_A |
402 |
232 |
180 |
2.17 |
18.89 |
56.069 |
7.942 |
T P |
| 2c1c_A |
312 |
232 |
181 |
2.11 |
20.44 |
55.956 |
8.198 |
T P |
| 3d68_A |
401 |
232 |
174 |
2.14 |
20.69 |
55.290 |
7.776 |
T P |
| 3d66_A |
401 |
232 |
172 |
2.13 |
21.51 |
54.964 |
7.702 |
T P |
| 1obr_A |
323 |
232 |
171 |
2.05 |
19.30 |
54.823 |
7.955 |
T P |
| 1zli_A |
306 |
232 |
174 |
2.08 |
17.24 |
54.297 |
7.980 |
T P |
| 3cdx_A |
329 |
232 |
166 |
1.97 |
21.08 |
53.522 |
8.031 |
T P |
| 1z5r_A |
304 |
232 |
174 |
2.11 |
17.82 |
53.486 |
7.889 |
T P |
| 3dgv_A |
401 |
232 |
172 |
2.08 |
20.93 |
53.437 |
7.904 |
T P |
| 3d4u_A |
296 |
232 |
169 |
2.09 |
20.12 |
53.391 |
7.711 |
T P |
| 2pcu_A |
305 |
232 |
174 |
2.14 |
20.11 |
53.158 |
7.781 |
T P |
| 1dtd_A |
303 |
232 |
176 |
2.16 |
17.61 |
52.849 |
7.787 |
T P |
| 1m4l_A |
307 |
232 |
176 |
2.17 |
17.61 |
52.667 |
7.750 |
T P |
| 2bk7_A |
307 |
232 |
171 |
2.13 |
19.88 |
52.370 |
7.663 |
T P |
| 1cbx_A |
307 |
232 |
177 |
2.19 |
18.08 |
52.146 |
7.731 |
T P |
| 5cpa_A |
307 |
232 |
176 |
2.16 |
17.61 |
52.103 |
7.788 |
T P |
| 1yme_A |
307 |
232 |
177 |
2.20 |
17.51 |
51.869 |
7.711 |
T P |
| 2ctc_A |
307 |
232 |
174 |
2.15 |
18.39 |
51.844 |
7.726 |
T P |
| 1aye_A |
401 |
232 |
176 |
2.18 |
17.61 |
51.637 |
7.721 |
T P |
| 1bav_A |
307 |
232 |
175 |
2.18 |
17.71 |
51.629 |
7.689 |
T P |
| 2v77_A |
308 |
232 |
175 |
2.17 |
18.86 |
51.510 |
7.710 |
T P |
| 3fju_A |
307 |
232 |
175 |
2.20 |
18.86 |
51.270 |
7.604 |
T P |
| 2g9d_A |
340 |
232 |
154 |
2.07 |
18.18 |
50.954 |
7.096 |
T P |
| 1pyt_B |
309 |
232 |
173 |
2.16 |
17.92 |
50.701 |
7.659 |
T P |
| 2boa_A |
404 |
232 |
167 |
2.14 |
20.96 |
49.816 |
7.464 |
T P |
| 2bco_A |
338 |
232 |
148 |
2.05 |
18.24 |
49.151 |
6.878 |
T P |
| 3fmc_C |
366 |
232 |
160 |
2.03 |
19.38 |
49.100 |
7.517 |
T P |
| 1yw4_A |
319 |
232 |
160 |
2.14 |
16.25 |
49.099 |
7.146 |
T P |
| 2qj8_A |
314 |
232 |
161 |
2.19 |
19.88 |
48.038 |
7.027 |
T P |
| 1yw6_A |
316 |
232 |
149 |
2.06 |
22.82 |
46.807 |
6.905 |
T P |
| 2o53_A |
302 |
232 |
145 |
2.07 |
19.31 |
44.164 |
6.667 |
T P |
| 2gu2_A |
307 |
232 |
146 |
2.09 |
19.18 |
44.134 |
6.664 |
T P |
| 2i3c_A |
302 |
232 |
143 |
2.08 |
18.88 |
43.139 |
6.569 |
T P |
| 1cpb_B |
217 |
232 |
119 |
2.20 |
18.49 |
36.203 |
5.174 |
T P |
| 1cpb_A |
82 |
232 |
42 |
2.65 |
7.14 |
11.865 |
1.525 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]