LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_135.5wLII_11080_4
Total number of 3D structures: 58
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
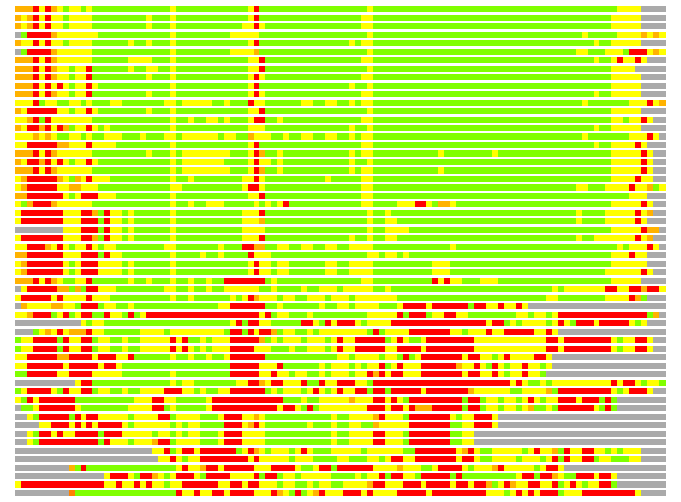
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2d29_A |
386 |
109 |
102 |
1.63 |
18.63 |
89.075 |
5.909 |
T P |
| 1egd_A |
387 |
109 |
102 |
1.71 |
12.75 |
88.588 |
5.650 |
T P |
| 2a1t_C |
388 |
109 |
102 |
1.71 |
12.75 |
88.126 |
5.647 |
T P |
| 2uxw_A |
567 |
109 |
104 |
1.84 |
14.42 |
87.991 |
5.361 |
T P |
| 1ege_A |
387 |
109 |
102 |
1.71 |
12.75 |
87.888 |
5.637 |
T P |
| 3b96_A |
554 |
109 |
101 |
1.69 |
14.85 |
87.321 |
5.653 |
T P |
| 2jif_A |
381 |
109 |
101 |
1.82 |
13.86 |
87.270 |
5.267 |
T P |
| 1jqi_A |
384 |
109 |
100 |
1.77 |
15.00 |
87.051 |
5.344 |
T P |
| 3mdd_A |
385 |
109 |
101 |
1.75 |
10.89 |
86.826 |
5.448 |
T P |
| 1ukw_A |
379 |
109 |
100 |
1.72 |
15.00 |
86.818 |
5.501 |
T P |
| 1udy_A |
385 |
109 |
101 |
1.78 |
10.89 |
86.432 |
5.382 |
T P |
| 1w07_B |
658 |
109 |
106 |
1.94 |
11.32 |
86.327 |
5.188 |
T P |
| 2vig_E |
380 |
109 |
98 |
1.54 |
16.33 |
86.154 |
5.964 |
T P |
| 2eba_A |
380 |
109 |
102 |
1.78 |
11.76 |
86.049 |
5.436 |
T P |
| 1rx0_A |
384 |
109 |
100 |
1.72 |
15.00 |
85.418 |
5.482 |
T P |
| 2fon_B |
656 |
109 |
107 |
2.08 |
12.15 |
85.394 |
4.904 |
T P |
| 2dvl_A |
370 |
109 |
98 |
1.73 |
16.33 |
83.735 |
5.360 |
T P |
| 2r0n_A |
390 |
109 |
103 |
2.06 |
9.71 |
83.577 |
4.771 |
T P |
| 1siq_A |
390 |
109 |
100 |
1.84 |
10.00 |
83.236 |
5.161 |
T P |
| 2r0m_A |
390 |
109 |
103 |
2.09 |
9.71 |
82.955 |
4.700 |
T P |
| 2pg0_A |
380 |
109 |
99 |
1.81 |
18.18 |
82.938 |
5.185 |
T P |
| 2z1q_B |
549 |
109 |
101 |
1.90 |
17.82 |
82.922 |
5.059 |
T P |
| 1buc_A |
383 |
109 |
96 |
1.54 |
15.62 |
82.641 |
5.841 |
T P |
| 3eom_A |
387 |
109 |
100 |
2.04 |
10.00 |
80.927 |
4.666 |
T P |
| 2c0u_A |
430 |
109 |
95 |
1.79 |
11.58 |
80.432 |
5.037 |
T P |
| 2c12_A |
430 |
109 |
94 |
1.68 |
11.70 |
80.235 |
5.294 |
T P |
| 2reh_A |
430 |
109 |
95 |
1.79 |
11.58 |
80.227 |
5.027 |
T P |
| 2zaf_A |
430 |
109 |
95 |
1.79 |
11.58 |
80.191 |
5.020 |
T P |
| 2ddh_A |
622 |
109 |
100 |
2.03 |
11.00 |
79.901 |
4.705 |
T P |
| 1ivh_A |
387 |
109 |
94 |
1.74 |
13.83 |
79.545 |
5.122 |
T P |
| 2ix6_A |
416 |
109 |
96 |
1.86 |
9.38 |
79.062 |
4.893 |
T P |
| 2ix5_A |
415 |
109 |
96 |
1.89 |
9.38 |
78.677 |
4.827 |
T P |
| 1r2j_A |
353 |
109 |
93 |
1.91 |
10.75 |
77.640 |
4.621 |
T P |
| 3djl_A |
538 |
109 |
95 |
2.16 |
13.68 |
74.047 |
4.209 |
T P |
| 2jbr_A |
399 |
109 |
97 |
2.17 |
7.22 |
67.941 |
4.275 |
T P |
| 2qr4_A |
521 |
109 |
62 |
2.23 |
9.68 |
44.239 |
2.659 |
T P |
| 3ce2_A |
595 |
109 |
59 |
2.18 |
10.17 |
42.231 |
2.592 |
T P |
| 1j36_A |
598 |
109 |
59 |
2.16 |
1.69 |
42.127 |
2.608 |
T P |
| 2h1n_A |
566 |
109 |
66 |
2.62 |
6.06 |
41.311 |
2.430 |
T P |
| 1afr_A |
345 |
109 |
66 |
2.63 |
1.52 |
41.212 |
2.422 |
T P |
| 2j2f_A |
348 |
109 |
64 |
2.75 |
4.69 |
41.022 |
2.249 |
T P |
| 2oc2_A |
583 |
109 |
58 |
2.37 |
1.72 |
40.969 |
2.352 |
T P |
| 3bkk_A |
586 |
109 |
60 |
2.37 |
3.33 |
40.805 |
2.428 |
T P |
| 2iul_A |
583 |
109 |
59 |
2.41 |
5.08 |
40.047 |
2.351 |
T P |
| 2iux_A |
577 |
109 |
61 |
2.26 |
3.28 |
39.891 |
2.586 |
T P |
| 2uw1_B |
338 |
109 |
61 |
2.63 |
1.64 |
39.836 |
2.235 |
T P |
| 2uw1_A |
327 |
109 |
58 |
2.39 |
3.45 |
38.677 |
2.325 |
T P |
| 1oq4_A |
346 |
109 |
58 |
2.46 |
1.72 |
38.413 |
2.265 |
T P |
| 1i1i_P |
665 |
109 |
55 |
2.51 |
5.45 |
37.854 |
2.103 |
T P |
| 2o3e_A |
665 |
109 |
54 |
2.53 |
5.56 |
37.281 |
2.052 |
T P |
| 1s4b_P |
654 |
109 |
53 |
2.43 |
1.89 |
36.414 |
2.094 |
T P |
| 2o36_A |
654 |
109 |
50 |
2.48 |
2.00 |
35.320 |
1.941 |
T P |
| 2ajf_A |
597 |
109 |
59 |
2.67 |
6.78 |
35.022 |
2.133 |
T P |
| 1r42_A |
597 |
109 |
56 |
2.46 |
7.14 |
34.547 |
2.186 |
T P |
| 1y79_1 |
680 |
109 |
55 |
2.68 |
3.64 |
34.514 |
1.981 |
T P |
| 2c6n_A |
612 |
109 |
53 |
2.50 |
0.00 |
34.150 |
2.038 |
T P |
| 3d0g_A |
597 |
109 |
54 |
2.80 |
5.56 |
33.939 |
1.859 |
T P |
| 1uze_A |
574 |
109 |
53 |
2.60 |
11.32 |
33.099 |
1.960 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]