LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_139.5wLII_11111_19
Total number of 3D structures: 110
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
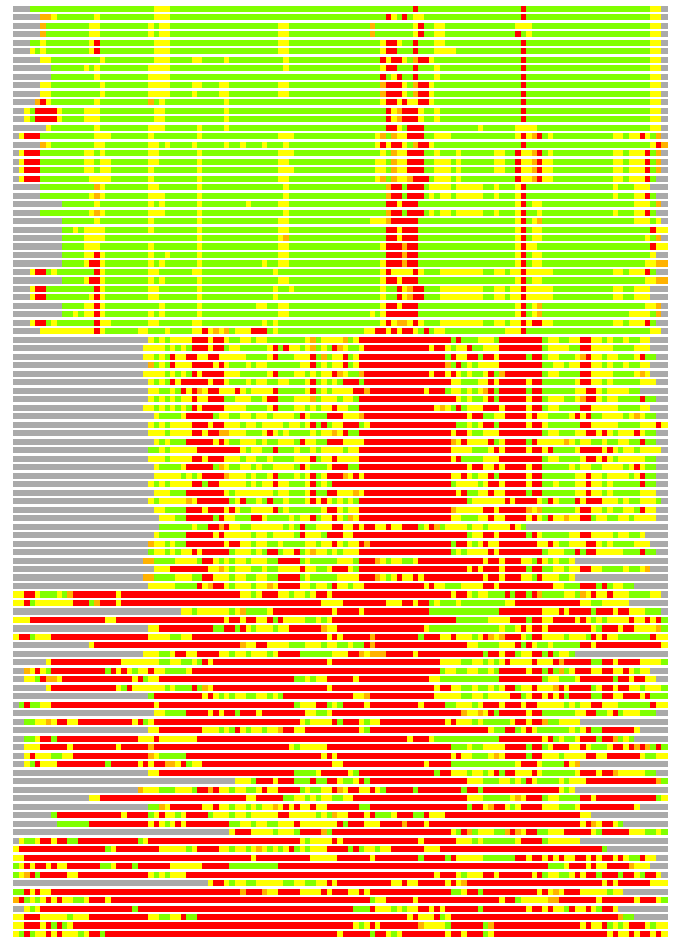
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1ehy_A |
282 |
121 |
115 |
0.67 |
15.65 |
94.418 |
14.905 |
T P |
| 1y37_A |
294 |
121 |
112 |
1.34 |
11.61 |
88.350 |
7.778 |
T P |
| 2cjp_A |
320 |
121 |
114 |
1.53 |
9.65 |
87.991 |
6.988 |
T P |
| 3cxu_A |
320 |
121 |
113 |
1.50 |
9.73 |
87.759 |
7.075 |
T P |
| 1zd3_A |
547 |
121 |
112 |
1.30 |
13.39 |
87.585 |
7.986 |
T P |
| 1cqz_B |
541 |
121 |
112 |
1.32 |
11.61 |
87.545 |
7.870 |
T P |
| 2og1_A |
285 |
121 |
109 |
1.39 |
11.93 |
85.776 |
7.292 |
T P |
| 2zjf_A |
346 |
121 |
109 |
1.25 |
11.01 |
85.769 |
8.084 |
T P |
| 2e3j_A |
346 |
121 |
109 |
1.23 |
11.93 |
85.766 |
8.184 |
T P |
| 2puj_A |
283 |
121 |
109 |
1.47 |
11.93 |
85.476 |
6.941 |
T P |
| 2rhw_A |
283 |
121 |
109 |
1.47 |
11.93 |
85.472 |
6.936 |
T P |
| 2vf2_A |
284 |
121 |
109 |
1.40 |
16.51 |
85.389 |
7.265 |
T P |
| 2d0d_A |
271 |
121 |
109 |
1.41 |
11.01 |
85.229 |
7.201 |
T P |
| 1iup_A |
271 |
121 |
109 |
1.42 |
11.01 |
85.033 |
7.166 |
T P |
| 1j1i_A |
258 |
121 |
109 |
1.45 |
12.84 |
84.660 |
7.037 |
T P |
| 1b6g_A |
308 |
121 |
110 |
1.74 |
11.82 |
84.321 |
5.971 |
T P |
| 1u2e_A |
286 |
121 |
109 |
1.55 |
16.51 |
84.120 |
6.596 |
T P |
| 2yxp_X |
310 |
121 |
110 |
1.78 |
12.73 |
83.743 |
5.836 |
T P |
| 1be0_A |
310 |
121 |
110 |
1.80 |
12.73 |
83.576 |
5.787 |
T P |
| 1bez_A |
310 |
121 |
110 |
1.79 |
12.73 |
83.507 |
5.822 |
T P |
| 1hde_A |
310 |
121 |
109 |
1.78 |
12.84 |
83.229 |
5.812 |
T P |
| 2v9z_A |
302 |
121 |
107 |
1.69 |
14.95 |
81.963 |
5.992 |
T P |
| 1cqw_A |
295 |
121 |
107 |
1.64 |
14.95 |
81.564 |
6.141 |
T P |
| 1a8s_A |
273 |
121 |
105 |
1.44 |
13.33 |
81.492 |
6.827 |
T P |
| 1bn6_A |
291 |
121 |
107 |
1.67 |
14.95 |
81.456 |
6.046 |
T P |
| 1a8q_A |
274 |
121 |
105 |
1.49 |
13.33 |
81.448 |
6.610 |
T P |
| 1a88_A |
275 |
121 |
105 |
1.47 |
9.52 |
81.311 |
6.703 |
T P |
| 1va4_A |
271 |
121 |
105 |
1.50 |
12.38 |
81.028 |
6.582 |
T P |
| 1zoi_A |
275 |
121 |
105 |
1.45 |
11.43 |
80.680 |
6.774 |
T P |
| 3fob_A |
277 |
121 |
103 |
1.47 |
17.48 |
80.437 |
6.575 |
T P |
| 1bro_A |
277 |
121 |
104 |
1.63 |
15.38 |
80.278 |
6.016 |
T P |
| 1cv2_A |
293 |
121 |
109 |
1.87 |
11.01 |
80.166 |
5.531 |
T P |
| 1brt_A |
277 |
121 |
104 |
1.60 |
15.38 |
80.140 |
6.110 |
T P |
| 1g42_A |
293 |
121 |
108 |
1.81 |
11.11 |
80.094 |
5.646 |
T P |
| 1iz7_A |
294 |
121 |
108 |
1.80 |
11.11 |
79.967 |
5.698 |
T P |
| 1a8u_A |
277 |
121 |
104 |
1.61 |
15.38 |
79.611 |
6.066 |
T P |
| 1hkh_A |
279 |
121 |
103 |
1.58 |
15.53 |
79.589 |
6.121 |
T P |
| 1mj5_A |
297 |
121 |
107 |
1.87 |
11.21 |
73.206 |
5.427 |
T P |
| 1wm1_A |
313 |
121 |
103 |
1.99 |
6.80 |
72.376 |
4.922 |
T P |
| 2ho3_D |
304 |
121 |
60 |
2.36 |
8.33 |
33.518 |
2.441 |
T P |
| 1evj_A |
340 |
121 |
61 |
2.42 |
6.56 |
33.513 |
2.422 |
T P |
| 1h6d_A |
383 |
121 |
57 |
2.29 |
8.77 |
32.507 |
2.385 |
T P |
| 2ho5_A |
305 |
121 |
58 |
2.40 |
6.90 |
32.479 |
2.320 |
T P |
| 1ofg_A |
381 |
121 |
62 |
2.51 |
6.45 |
32.311 |
2.374 |
T P |
| 3e9m_A |
321 |
121 |
56 |
2.27 |
12.50 |
32.033 |
2.358 |
T P |
| 2ixa_A |
426 |
121 |
59 |
2.62 |
3.39 |
32.022 |
2.170 |
T P |
| 3db2_A |
347 |
121 |
59 |
2.38 |
6.78 |
31.999 |
2.379 |
T P |
| 1ryd_A |
381 |
121 |
61 |
2.48 |
6.56 |
31.898 |
2.365 |
T P |
| 3f4l_A |
344 |
121 |
56 |
2.39 |
3.57 |
31.675 |
2.251 |
T P |
| 2nvw_A |
413 |
121 |
58 |
2.34 |
15.52 |
31.210 |
2.374 |
T P |
| 1lc0_A |
290 |
121 |
57 |
2.59 |
3.51 |
31.197 |
2.122 |
T P |
| 3ec7_A |
336 |
121 |
59 |
2.51 |
11.86 |
31.132 |
2.260 |
T P |
| 3euw_A |
333 |
121 |
56 |
2.49 |
16.07 |
31.034 |
2.159 |
T P |
| 1zh8_A |
325 |
121 |
58 |
2.43 |
6.90 |
30.992 |
2.294 |
T P |
| 2h63_D |
285 |
121 |
54 |
2.37 |
12.96 |
30.836 |
2.184 |
T P |
| 3c1a_A |
307 |
121 |
56 |
2.47 |
12.50 |
30.665 |
2.183 |
T P |
| 2o4u_X |
331 |
121 |
56 |
2.51 |
8.93 |
30.634 |
2.146 |
T P |
| 3fhl_C |
338 |
121 |
57 |
2.57 |
10.53 |
30.456 |
2.133 |
T P |
| 1tlt_B |
305 |
121 |
55 |
2.44 |
10.91 |
30.304 |
2.168 |
T P |
| 1xea_A |
311 |
121 |
56 |
2.49 |
12.50 |
30.241 |
2.165 |
T P |
| 2glx_A |
332 |
121 |
56 |
2.59 |
10.71 |
30.219 |
2.081 |
T P |
| 3btu_A |
392 |
121 |
53 |
2.48 |
3.77 |
30.170 |
2.056 |
T P |
| 1gcu_A |
292 |
121 |
50 |
2.43 |
14.00 |
29.402 |
1.977 |
T P |
| 1ydw_B |
350 |
121 |
54 |
2.52 |
3.70 |
29.242 |
2.062 |
T P |
| 3e82_D |
350 |
121 |
55 |
2.46 |
14.55 |
29.079 |
2.148 |
T P |
| 1p4c_A |
353 |
121 |
53 |
2.59 |
7.55 |
28.520 |
1.968 |
T P |
| 3e18_A |
348 |
121 |
49 |
2.53 |
10.20 |
28.199 |
1.866 |
T P |
| 2a7n_A |
353 |
121 |
52 |
2.58 |
5.77 |
28.191 |
1.941 |
T P |
| 3dty_A |
374 |
121 |
54 |
2.55 |
7.41 |
28.191 |
2.036 |
T P |
| 1kbi_A |
504 |
121 |
53 |
2.51 |
11.32 |
27.919 |
2.033 |
T P |
| 2cu0_A |
358 |
121 |
46 |
2.51 |
2.17 |
26.453 |
1.765 |
T P |
| 2z6j_A |
307 |
121 |
44 |
2.32 |
4.55 |
26.322 |
1.818 |
T P |
| 1gox_A |
350 |
121 |
45 |
2.60 |
6.67 |
26.182 |
1.667 |
T P |
| 2c6q_B |
329 |
121 |
45 |
2.42 |
11.11 |
26.130 |
1.785 |
T P |
| 2oz0_A |
492 |
121 |
49 |
2.67 |
10.20 |
25.867 |
1.768 |
T P |
| 1ypf_A |
295 |
121 |
44 |
2.37 |
13.64 |
25.582 |
1.780 |
T P |
| 1tb3_A |
332 |
121 |
44 |
2.64 |
6.82 |
25.263 |
1.606 |
T P |
| 1nf7_A |
454 |
121 |
46 |
2.70 |
6.52 |
25.189 |
1.640 |
T P |
| 2cdh_0 |
226 |
121 |
42 |
2.53 |
11.90 |
25.066 |
1.599 |
T P |
| 2zfa_A |
352 |
121 |
41 |
2.38 |
7.32 |
24.893 |
1.655 |
T P |
| 1jr1_A |
436 |
121 |
47 |
2.74 |
8.51 |
24.824 |
1.652 |
T P |
| 3cea_A |
342 |
121 |
45 |
2.59 |
11.11 |
24.296 |
1.674 |
T P |
| 1ltd_A |
481 |
121 |
45 |
2.58 |
8.89 |
24.188 |
1.679 |
T P |
| 3ezy_A |
334 |
121 |
40 |
2.29 |
10.00 |
24.162 |
1.672 |
T P |
| 2rdu_A |
360 |
121 |
42 |
2.53 |
7.14 |
24.095 |
1.596 |
T P |
| 2p2s_A |
333 |
121 |
38 |
2.57 |
5.26 |
23.914 |
1.422 |
T P |
| 2nli_A |
352 |
121 |
39 |
2.34 |
12.82 |
23.815 |
1.598 |
T P |
| 2nzl_A |
339 |
121 |
38 |
2.69 |
7.89 |
23.356 |
1.364 |
T P |
| 3fd8_D |
349 |
121 |
42 |
2.64 |
11.90 |
22.865 |
1.534 |
T P |
| 1szf_A |
395 |
121 |
37 |
2.44 |
5.41 |
22.809 |
1.455 |
T P |
| 3btv_B |
392 |
121 |
39 |
2.42 |
5.13 |
22.669 |
1.550 |
T P |
| 1qcw_A |
385 |
121 |
38 |
2.29 |
10.53 |
22.559 |
1.591 |
T P |
| 2j6x_A |
370 |
121 |
40 |
2.64 |
2.50 |
22.413 |
1.460 |
T P |
| 1eep_A |
314 |
121 |
38 |
2.45 |
2.63 |
22.351 |
1.490 |
T P |
| 3evn_A |
316 |
121 |
39 |
2.67 |
2.56 |
22.298 |
1.407 |
T P |
| 2z6i_B |
321 |
121 |
44 |
2.81 |
6.82 |
22.220 |
1.514 |
T P |
| 2gjl_A |
324 |
121 |
41 |
2.73 |
4.88 |
22.023 |
1.448 |
T P |
| 1vrd_A |
321 |
121 |
38 |
2.75 |
7.89 |
21.728 |
1.331 |
T P |
| 1lco_A |
480 |
121 |
36 |
2.44 |
5.56 |
21.710 |
1.418 |
T P |
| 2a7r_A |
338 |
121 |
40 |
2.64 |
5.00 |
21.008 |
1.461 |
T P |
| 1al8_A |
344 |
121 |
35 |
2.67 |
5.71 |
20.610 |
1.263 |
T P |
| 3e1k_A |
395 |
121 |
36 |
2.56 |
2.78 |
20.326 |
1.352 |
T P |
| 1kbj_A |
393 |
121 |
33 |
2.60 |
3.03 |
18.864 |
1.222 |
T P |
| 1zfj_A |
476 |
121 |
33 |
2.77 |
6.06 |
17.914 |
1.148 |
T P |
| 1sze_A |
392 |
121 |
33 |
2.98 |
12.12 |
17.685 |
1.073 |
T P |
| 3bw3_A |
346 |
121 |
29 |
2.75 |
3.45 |
16.606 |
1.016 |
T P |
| 3bw4_A |
347 |
121 |
28 |
2.59 |
10.71 |
16.108 |
1.041 |
T P |
| 3bo9_A |
314 |
121 |
28 |
2.80 |
7.14 |
15.248 |
0.965 |
T P |
| 1gyl_A |
352 |
121 |
29 |
2.89 |
10.34 |
15.043 |
0.971 |
T P |
| 3bw2_A |
347 |
121 |
27 |
2.83 |
18.52 |
14.651 |
0.921 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]