LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_145.5wLII_11111_49
Total number of 3D structures: 72
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
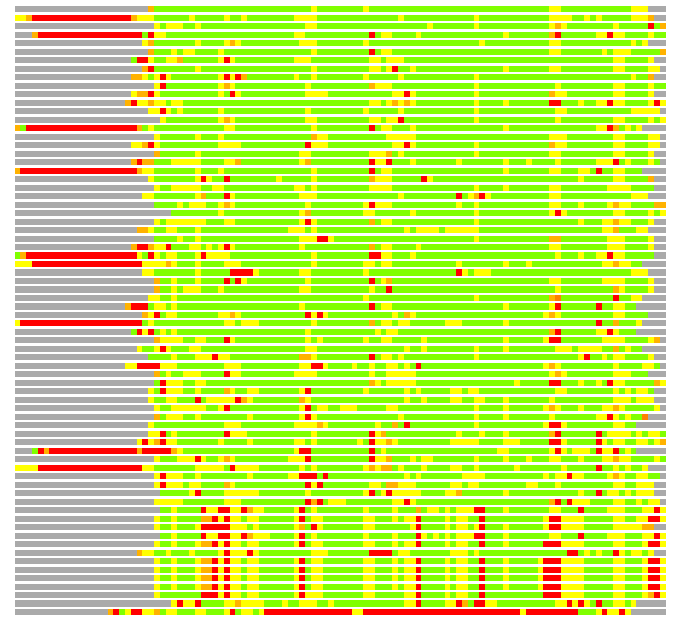
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2nzi_A |
292 |
112 |
86 |
1.06 |
19.77 |
75.094 |
7.415 |
T P |
| 2yux_A |
120 |
112 |
93 |
1.93 |
21.51 |
75.050 |
4.581 |
T P |
| 1uem_A |
117 |
112 |
87 |
1.54 |
21.84 |
73.363 |
5.320 |
T P |
| 1tdq_A |
271 |
112 |
87 |
1.62 |
17.24 |
73.163 |
5.056 |
T P |
| 1x4z_A |
121 |
112 |
87 |
1.60 |
19.54 |
73.085 |
5.108 |
T P |
| 1x5l_A |
111 |
112 |
88 |
1.68 |
10.23 |
72.921 |
4.948 |
T P |
| 2edx_A |
134 |
112 |
87 |
1.69 |
16.09 |
72.791 |
4.862 |
T P |
| 2doc_A |
119 |
112 |
87 |
1.55 |
10.34 |
72.787 |
5.279 |
T P |
| 1wf5_A |
121 |
112 |
87 |
1.72 |
13.79 |
72.274 |
4.775 |
T P |
| 3f7r_A |
214 |
112 |
87 |
1.58 |
10.34 |
72.145 |
5.177 |
T P |
| 1i1r_A |
301 |
112 |
87 |
1.79 |
16.09 |
72.086 |
4.601 |
T P |
| 1fnf_A |
368 |
112 |
88 |
1.90 |
15.91 |
72.022 |
4.400 |
T P |
| 1bpv_A |
104 |
112 |
87 |
1.55 |
20.69 |
71.973 |
5.286 |
T P |
| 3f7p_E |
212 |
112 |
86 |
1.50 |
11.63 |
71.793 |
5.388 |
T P |
| 1qg3_A |
195 |
112 |
87 |
1.85 |
11.49 |
71.448 |
4.472 |
T P |
| 1x5f_A |
120 |
112 |
87 |
1.70 |
12.64 |
71.315 |
4.838 |
T P |
| 1bqu_B |
215 |
112 |
88 |
1.88 |
15.91 |
71.166 |
4.448 |
T P |
| 2dju_A |
106 |
112 |
86 |
1.64 |
23.26 |
71.053 |
4.933 |
T P |
| 1x5k_A |
124 |
112 |
87 |
1.95 |
20.69 |
70.939 |
4.251 |
T P |
| 1wfo_A |
130 |
112 |
86 |
1.75 |
16.28 |
70.802 |
4.643 |
T P |
| 2vkw_A |
197 |
112 |
84 |
1.60 |
16.67 |
70.685 |
4.929 |
T P |
| 2djs_A |
108 |
112 |
86 |
1.65 |
18.60 |
70.610 |
4.906 |
T P |
| 1cfb_A |
205 |
112 |
84 |
1.60 |
11.90 |
70.293 |
4.953 |
T P |
| 1x5g_A |
116 |
112 |
87 |
1.88 |
19.54 |
70.254 |
4.392 |
T P |
| 2uvf_A |
571 |
112 |
84 |
1.57 |
21.43 |
70.168 |
5.015 |
T P |
| 1x5h_A |
132 |
112 |
85 |
1.75 |
11.76 |
70.105 |
4.606 |
T P |
| 1x3d_A |
118 |
112 |
88 |
1.89 |
17.05 |
70.101 |
4.412 |
T P |
| 1x5x_A |
109 |
112 |
84 |
1.68 |
20.24 |
70.081 |
4.732 |
T P |
| 1va9_A |
122 |
112 |
86 |
1.84 |
12.79 |
70.075 |
4.441 |
T P |
| 2v5y_A |
564 |
112 |
86 |
1.85 |
17.44 |
70.026 |
4.413 |
T P |
| 2dlh_A |
121 |
112 |
89 |
1.93 |
12.36 |
69.872 |
4.385 |
T P |
| 1uey_A |
127 |
112 |
82 |
1.50 |
17.07 |
69.854 |
5.126 |
T P |
| 2ede_A |
114 |
112 |
83 |
1.60 |
21.69 |
69.799 |
4.877 |
T P |
| 2edy_A |
103 |
112 |
85 |
1.65 |
12.94 |
69.796 |
4.846 |
T P |
| 1k85_A |
88 |
112 |
84 |
1.61 |
16.67 |
69.615 |
4.909 |
T P |
| 1qr4_A |
175 |
112 |
82 |
1.51 |
14.63 |
69.612 |
5.092 |
T P |
| 1pvh_A |
201 |
112 |
84 |
1.82 |
16.67 |
69.490 |
4.374 |
T P |
| 3f7q_A |
214 |
112 |
86 |
1.91 |
12.79 |
69.385 |
4.284 |
T P |
| 1fnh_A |
269 |
112 |
83 |
1.55 |
14.46 |
69.290 |
5.040 |
T P |
| 2ee2_A |
119 |
112 |
84 |
1.74 |
14.29 |
69.212 |
4.558 |
T P |
| 2w1n_A |
228 |
112 |
86 |
1.77 |
22.09 |
69.173 |
4.595 |
T P |
| 2dmk_A |
127 |
112 |
84 |
1.78 |
19.05 |
69.139 |
4.456 |
T P |
| 2q7n_A |
480 |
112 |
85 |
1.80 |
11.76 |
68.941 |
4.474 |
T P |
| 1uen_A |
125 |
112 |
84 |
1.82 |
14.29 |
68.781 |
4.372 |
T P |
| 1mfn_A |
184 |
112 |
84 |
1.80 |
17.86 |
68.781 |
4.411 |
T P |
| 1x4x_A |
106 |
112 |
85 |
1.87 |
17.65 |
68.260 |
4.305 |
T P |
| 2db8_A |
110 |
112 |
85 |
1.90 |
15.29 |
68.172 |
4.253 |
T P |
| 1x5z_A |
115 |
112 |
85 |
1.90 |
20.00 |
67.932 |
4.250 |
T P |
| 2yuw_A |
110 |
112 |
83 |
1.82 |
15.66 |
67.422 |
4.329 |
T P |
| 1p9m_A |
298 |
112 |
81 |
1.73 |
17.28 |
66.667 |
4.427 |
T P |
| 2gee_A |
188 |
112 |
84 |
1.84 |
17.86 |
66.172 |
4.322 |
T P |
| 2dkm_A |
104 |
112 |
85 |
2.07 |
24.71 |
64.515 |
3.915 |
T P |
| 2qbw_A |
189 |
112 |
77 |
1.78 |
15.58 |
63.421 |
4.101 |
T P |
| 2ed8_A |
106 |
112 |
85 |
1.98 |
14.12 |
63.419 |
4.082 |
T P |
| 2vkx_E |
202 |
112 |
89 |
2.11 |
12.36 |
62.205 |
4.023 |
T P |
| 1wfu_A |
120 |
112 |
81 |
1.89 |
16.05 |
62.001 |
4.063 |
T P |
| 2ed9_A |
124 |
112 |
83 |
2.03 |
13.25 |
61.130 |
3.897 |
T P |
| 2dn7_A |
107 |
112 |
81 |
2.00 |
17.28 |
60.314 |
3.857 |
T P |
| 1bj8_A |
109 |
112 |
82 |
1.95 |
15.85 |
57.782 |
4.005 |
T P |
| 3b8s_A |
567 |
112 |
78 |
2.07 |
16.67 |
56.083 |
3.592 |
T P |
| 1ctn_A |
538 |
112 |
79 |
2.04 |
8.86 |
55.988 |
3.692 |
T P |
| 1x6l_A |
539 |
112 |
76 |
2.04 |
9.21 |
55.141 |
3.546 |
T P |
| 3b9e_A |
581 |
112 |
77 |
2.05 |
16.88 |
54.948 |
3.582 |
T P |
| 1edq_A |
540 |
112 |
78 |
2.00 |
7.69 |
54.569 |
3.711 |
T P |
| 3fl7_A |
482 |
112 |
80 |
1.93 |
16.25 |
54.547 |
3.950 |
T P |
| 1k9t_A |
538 |
112 |
78 |
2.01 |
7.69 |
54.345 |
3.688 |
T P |
| 1ehn_A |
540 |
112 |
78 |
2.01 |
7.69 |
54.311 |
3.703 |
T P |
| 1eib_A |
540 |
112 |
78 |
2.01 |
7.69 |
54.292 |
3.693 |
T P |
| 1ffr_A |
540 |
112 |
78 |
2.02 |
7.69 |
53.652 |
3.676 |
T P |
| 1nh6_A |
540 |
112 |
77 |
2.06 |
7.79 |
52.856 |
3.573 |
T P |
| 1v8h_A |
106 |
112 |
71 |
2.15 |
18.31 |
49.673 |
3.149 |
T P |
| 1cz1_A |
394 |
112 |
33 |
2.61 |
3.03 |
20.684 |
1.218 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]