LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_146.5wLII_11111_54
Total number of 3D structures: 59
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
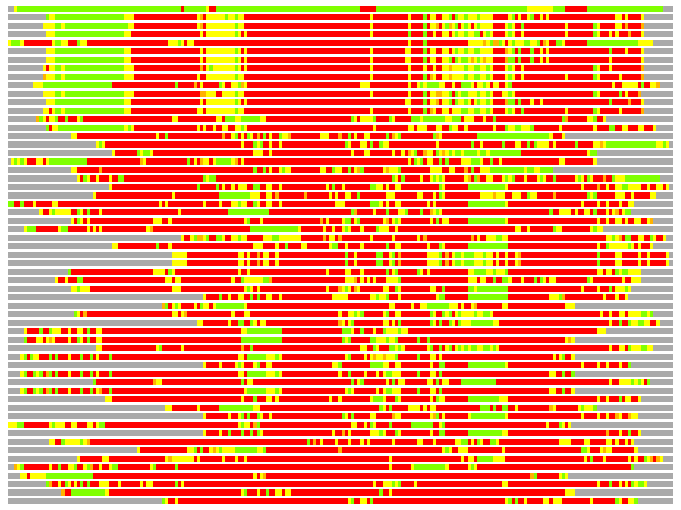
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3b4r_A |
218 |
211 |
191 |
0.90 |
23.04 |
88.896 |
19.098 |
T P |
| 3ejy_A |
444 |
211 |
73 |
2.48 |
6.85 |
23.657 |
2.825 |
T P |
| 2ht2_A |
444 |
211 |
74 |
2.61 |
6.76 |
23.450 |
2.726 |
T P |
| 2ez0_A |
444 |
211 |
69 |
2.42 |
5.80 |
23.417 |
2.742 |
T P |
| 1otu_A |
444 |
211 |
70 |
2.36 |
7.14 |
23.396 |
2.847 |
T P |
| 2htk_A |
444 |
211 |
71 |
2.50 |
4.23 |
23.208 |
2.731 |
T P |
| 2exw_A |
444 |
211 |
70 |
2.52 |
4.29 |
23.098 |
2.673 |
T P |
| 1ott_A |
444 |
211 |
69 |
2.44 |
4.35 |
22.980 |
2.720 |
T P |
| 2ht4_A |
444 |
211 |
72 |
2.49 |
6.94 |
22.971 |
2.778 |
T P |
| 3det_A |
444 |
211 |
71 |
2.39 |
8.45 |
22.963 |
2.847 |
T P |
| 2htl_A |
444 |
211 |
72 |
2.53 |
5.56 |
22.876 |
2.740 |
T P |
| 2ht3_A |
444 |
211 |
69 |
2.51 |
4.35 |
22.744 |
2.643 |
T P |
| 2fed_A |
444 |
211 |
70 |
2.48 |
7.14 |
22.622 |
2.717 |
T P |
| 2exy_A |
444 |
211 |
62 |
2.31 |
8.06 |
22.365 |
2.574 |
T P |
| 1kpl_B |
451 |
211 |
67 |
2.43 |
4.48 |
21.652 |
2.649 |
T P |
| 2cu0_A |
358 |
211 |
57 |
2.51 |
5.26 |
19.605 |
2.188 |
T P |
| 3ddj_A |
279 |
211 |
55 |
2.36 |
5.45 |
18.647 |
2.234 |
T P |
| 1jcn_A |
395 |
211 |
51 |
2.48 |
5.88 |
18.519 |
1.976 |
T P |
| 2qlv_C |
310 |
211 |
56 |
2.41 |
7.14 |
18.427 |
2.228 |
T P |
| 1eep_A |
314 |
211 |
56 |
2.63 |
5.36 |
18.268 |
2.052 |
T P |
| 2oux_A |
257 |
211 |
56 |
2.31 |
3.57 |
18.235 |
2.324 |
T P |
| 2yvx_A |
442 |
211 |
53 |
2.49 |
3.77 |
17.745 |
2.046 |
T P |
| 1zfj_A |
476 |
211 |
56 |
2.72 |
10.71 |
17.556 |
1.987 |
T P |
| 2v8q_E |
304 |
211 |
51 |
2.51 |
5.88 |
17.032 |
1.954 |
T P |
| 1pvm_B |
184 |
211 |
53 |
2.49 |
3.77 |
16.844 |
2.044 |
T P |
| 2o16_B |
135 |
211 |
49 |
2.55 |
10.20 |
16.644 |
1.848 |
T P |
| 1nf7_A |
454 |
211 |
49 |
2.46 |
2.04 |
16.544 |
1.913 |
T P |
| 1jr1_A |
436 |
211 |
56 |
3.04 |
3.57 |
16.210 |
1.784 |
T P |
| 2j9l_C |
172 |
211 |
47 |
2.42 |
8.51 |
16.140 |
1.868 |
T P |
| 1pvn_A |
362 |
211 |
48 |
2.70 |
6.25 |
15.855 |
1.714 |
T P |
| 1mwf_A |
362 |
211 |
48 |
2.70 |
6.25 |
15.855 |
1.714 |
T P |
| 2ef7_A |
127 |
211 |
44 |
2.49 |
11.36 |
15.785 |
1.701 |
T P |
| 2yvy_A |
248 |
211 |
52 |
2.77 |
1.92 |
15.714 |
1.811 |
T P |
| 3fna_B |
123 |
211 |
48 |
2.44 |
4.17 |
15.631 |
1.887 |
T P |
| 2qrd_G |
325 |
211 |
46 |
2.54 |
13.04 |
15.466 |
1.745 |
T P |
| 3ctu_B |
152 |
211 |
43 |
2.48 |
6.98 |
15.383 |
1.667 |
T P |
| 2p9m_B |
129 |
211 |
49 |
2.67 |
2.04 |
15.256 |
1.768 |
T P |
| 1y5h_A |
123 |
211 |
45 |
2.43 |
2.22 |
15.194 |
1.780 |
T P |
| 2yzi_B |
136 |
211 |
42 |
2.28 |
4.76 |
15.182 |
1.762 |
T P |
| 1xkf_B |
123 |
211 |
42 |
2.30 |
9.52 |
15.138 |
1.747 |
T P |
| 1pbj_A |
120 |
211 |
46 |
2.49 |
8.70 |
14.715 |
1.773 |
T P |
| 2uv4_A |
143 |
211 |
45 |
2.48 |
8.89 |
14.659 |
1.746 |
T P |
| 2rc3_C |
128 |
211 |
41 |
2.30 |
2.44 |
14.541 |
1.705 |
T P |
| 2uv7_A |
143 |
211 |
44 |
2.42 |
13.64 |
14.418 |
1.744 |
T P |
| 1vr9_A |
121 |
211 |
40 |
2.35 |
7.50 |
14.320 |
1.634 |
T P |
| 2uv6_A |
143 |
211 |
44 |
2.51 |
11.36 |
14.162 |
1.685 |
T P |
| 2nyc_A |
130 |
211 |
39 |
2.36 |
2.56 |
13.920 |
1.587 |
T P |
| 3fhm_A |
136 |
211 |
41 |
2.35 |
4.88 |
13.873 |
1.675 |
T P |
| 2rih_A |
131 |
211 |
41 |
2.52 |
12.20 |
13.742 |
1.566 |
T P |
| 2emq_A |
135 |
211 |
42 |
2.49 |
9.52 |
13.429 |
1.624 |
T P |
| 2nye_A |
132 |
211 |
37 |
2.42 |
2.70 |
13.403 |
1.467 |
T P |
| 2yzq_A |
224 |
211 |
43 |
2.89 |
4.65 |
13.145 |
1.439 |
T P |
| 2oox_E |
333 |
211 |
39 |
2.58 |
2.56 |
13.068 |
1.458 |
T P |
| 2yvz_A |
248 |
211 |
39 |
2.98 |
10.26 |
12.687 |
1.266 |
T P |
| 1o50_A |
141 |
211 |
35 |
2.35 |
0.00 |
12.392 |
1.426 |
T P |
| 3ejz_A |
444 |
211 |
30 |
2.33 |
10.00 |
11.576 |
1.233 |
T P |
| 2qh1_B |
185 |
211 |
36 |
2.55 |
8.33 |
10.950 |
1.358 |
T P |
| 3fio_A |
70 |
211 |
26 |
2.18 |
0.00 |
10.209 |
1.140 |
T P |
| 1ots_A |
444 |
211 |
23 |
2.81 |
13.04 |
7.939 |
0.792 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]