LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_150.5wLII_11111_90
Total number of 3D structures: 58
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
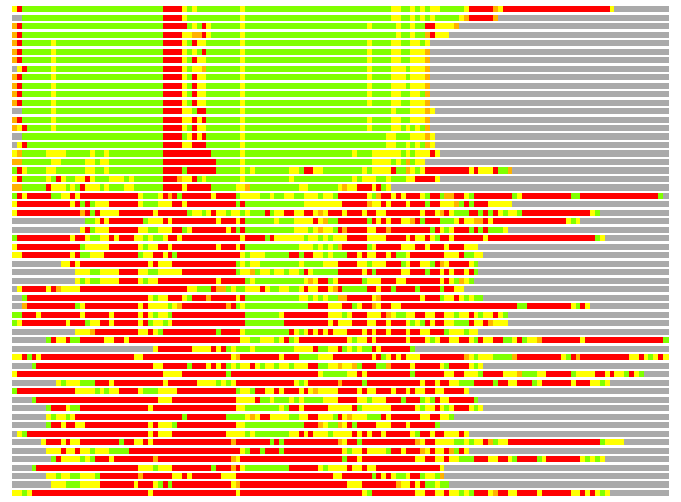
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1jhw_A |
326 |
135 |
92 |
1.63 |
16.30 |
65.322 |
5.323 |
T P |
| 1j72_A |
337 |
135 |
89 |
1.32 |
16.85 |
64.682 |
6.275 |
T P |
| 2fgh_A |
721 |
135 |
83 |
1.59 |
8.43 |
58.811 |
4.902 |
T P |
| 1d0n_A |
729 |
135 |
83 |
1.73 |
8.43 |
58.031 |
4.547 |
T P |
| 1p8z_G |
131 |
135 |
80 |
1.45 |
8.75 |
56.676 |
5.157 |
T P |
| 1d4x_G |
124 |
135 |
80 |
1.46 |
8.75 |
56.618 |
5.113 |
T P |
| 1c0g_S |
127 |
135 |
80 |
1.47 |
8.75 |
56.524 |
5.101 |
T P |
| 3cip_G |
128 |
135 |
80 |
1.51 |
8.75 |
56.472 |
4.977 |
T P |
| 1nlv_G |
123 |
135 |
80 |
1.47 |
8.75 |
56.465 |
5.104 |
T P |
| 3ci5_G |
126 |
135 |
80 |
1.46 |
8.75 |
56.410 |
5.113 |
T P |
| 1esv_S |
125 |
135 |
80 |
1.48 |
8.75 |
56.407 |
5.051 |
T P |
| 2ff3_A |
125 |
135 |
80 |
1.47 |
8.75 |
56.352 |
5.088 |
T P |
| 1db0_B |
317 |
135 |
79 |
1.31 |
8.86 |
56.316 |
5.586 |
T P |
| 1t44_G |
144 |
135 |
79 |
1.51 |
8.86 |
56.270 |
4.908 |
T P |
| 1rgi_G |
346 |
135 |
80 |
1.48 |
8.75 |
56.183 |
5.064 |
T P |
| 1h1v_G |
327 |
135 |
79 |
1.34 |
8.86 |
56.183 |
5.468 |
T P |
| 2fh1_B |
324 |
135 |
78 |
1.37 |
8.97 |
55.523 |
5.294 |
T P |
| 1svy_A |
102 |
135 |
77 |
1.91 |
10.39 |
50.861 |
3.838 |
T P |
| 1kcq_A |
103 |
135 |
74 |
1.75 |
13.51 |
50.832 |
4.004 |
T P |
| 1nph_A |
323 |
135 |
78 |
1.96 |
11.54 |
49.938 |
3.779 |
T P |
| 1vil |
126 |
135 |
77 |
1.93 |
11.69 |
47.759 |
3.792 |
T P |
| 1svq_A |
94 |
135 |
63 |
2.24 |
11.11 |
38.203 |
2.688 |
T P |
| 1fuu_B |
380 |
135 |
53 |
2.59 |
11.32 |
27.635 |
1.970 |
T P |
| 1s2m_A |
377 |
135 |
53 |
2.62 |
13.21 |
26.951 |
1.951 |
T P |
| 2vso_A |
366 |
135 |
51 |
2.72 |
7.84 |
25.349 |
1.807 |
T P |
| 3ews_A |
416 |
135 |
48 |
2.70 |
10.42 |
25.344 |
1.713 |
T P |
| 2va8_B |
695 |
135 |
49 |
2.40 |
8.16 |
25.108 |
1.958 |
T P |
| 2i4i_A |
408 |
135 |
46 |
2.68 |
10.87 |
23.218 |
1.656 |
T P |
| 1hv8_A |
363 |
135 |
44 |
2.73 |
18.18 |
23.186 |
1.557 |
T P |
| 2hyi_C |
392 |
135 |
45 |
2.60 |
0.00 |
22.860 |
1.669 |
T P |
| 1c4o_A |
504 |
135 |
43 |
2.50 |
4.65 |
22.716 |
1.652 |
T P |
| 2j0s_A |
391 |
135 |
47 |
2.66 |
10.64 |
22.647 |
1.705 |
T P |
| 2z0m_A |
331 |
135 |
43 |
2.58 |
4.65 |
22.547 |
1.602 |
T P |
| 2j0u_A |
366 |
135 |
44 |
2.73 |
11.36 |
22.378 |
1.556 |
T P |
| 2db3_A |
420 |
135 |
41 |
2.48 |
4.88 |
22.289 |
1.588 |
T P |
| 1wp9_A |
479 |
135 |
41 |
2.53 |
9.76 |
21.956 |
1.558 |
T P |
| 1oyw_A |
516 |
135 |
46 |
2.51 |
4.35 |
21.944 |
1.766 |
T P |
| 1oyy_A |
512 |
135 |
44 |
2.61 |
6.82 |
21.598 |
1.623 |
T P |
| 3g0h_A |
408 |
135 |
40 |
2.63 |
10.00 |
21.425 |
1.467 |
T P |
| 2hxy_A |
376 |
135 |
43 |
2.76 |
11.63 |
21.330 |
1.504 |
T P |
| 2rb4_A |
164 |
135 |
36 |
2.32 |
13.89 |
20.596 |
1.487 |
T P |
| 1gm5_A |
729 |
135 |
43 |
2.87 |
0.00 |
20.513 |
1.447 |
T P |
| 2zu6_C |
351 |
135 |
40 |
2.57 |
2.50 |
20.430 |
1.498 |
T P |
| 2eyq_A |
1146 |
135 |
42 |
2.66 |
0.00 |
20.276 |
1.524 |
T P |
| 1d9x_A |
590 |
135 |
40 |
2.64 |
10.00 |
19.950 |
1.462 |
T P |
| 3fht_A |
392 |
135 |
42 |
2.89 |
9.52 |
19.624 |
1.407 |
T P |
| 1d2m_A |
552 |
135 |
37 |
2.46 |
2.70 |
19.366 |
1.444 |
T P |
| 1d9z_A |
590 |
135 |
39 |
2.69 |
5.13 |
19.102 |
1.399 |
T P |
| 2jgn_B |
161 |
135 |
40 |
2.65 |
2.50 |
19.032 |
1.453 |
T P |
| 2g2j_A |
158 |
135 |
35 |
2.48 |
2.86 |
18.945 |
1.357 |
T P |
| 2fwr_A |
434 |
135 |
40 |
2.79 |
10.00 |
18.903 |
1.384 |
T P |
| 1t5l_A |
595 |
135 |
35 |
2.90 |
5.71 |
17.600 |
1.166 |
T P |
| 3eaq_B |
209 |
135 |
36 |
2.66 |
13.89 |
16.980 |
1.306 |
T P |
| 2d7d_A |
621 |
135 |
34 |
2.72 |
8.82 |
16.373 |
1.205 |
T P |
| 2fzl_A |
197 |
135 |
31 |
2.58 |
6.45 |
16.201 |
1.156 |
T P |
| 2hjv_A |
158 |
135 |
31 |
2.82 |
3.23 |
15.691 |
1.063 |
T P |
| 1fuk_A |
157 |
135 |
28 |
2.91 |
7.14 |
13.876 |
0.930 |
T P |
| 2fdc_B |
585 |
135 |
28 |
2.96 |
3.57 |
12.681 |
0.914 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]