LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_168.5wLII_11172_54
Total number of 3D structures: 30
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
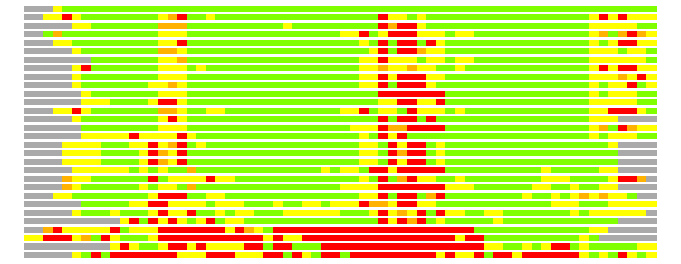
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1xbl_A |
75 |
66 |
63 |
0.55 |
34.92 |
94.996 |
9.746 |
T P |
| 2qsa_A |
101 |
66 |
59 |
1.71 |
28.81 |
84.243 |
3.259 |
T P |
| 2dmx_A |
92 |
66 |
58 |
1.83 |
34.48 |
81.165 |
3.005 |
T P |
| 1wjz_A |
94 |
66 |
59 |
2.03 |
18.64 |
81.043 |
2.770 |
T P |
| 2cug_A |
88 |
66 |
56 |
1.59 |
30.36 |
80.912 |
3.304 |
T P |
| 2ej7_A |
82 |
66 |
58 |
1.83 |
29.31 |
80.649 |
2.998 |
T P |
| 2ctw_A |
109 |
66 |
58 |
1.86 |
27.59 |
80.464 |
2.958 |
T P |
| 2dn9_A |
79 |
66 |
57 |
1.89 |
29.82 |
79.150 |
2.868 |
T P |
| 2ctp_A |
78 |
66 |
56 |
1.87 |
25.00 |
78.785 |
2.837 |
T P |
| 2ctr_A |
88 |
66 |
56 |
1.78 |
32.14 |
78.264 |
2.975 |
T P |
| 2o37_A |
81 |
66 |
53 |
1.52 |
28.30 |
77.592 |
3.278 |
T P |
| 2ctq_A |
112 |
66 |
55 |
1.74 |
20.00 |
76.586 |
2.991 |
T P |
| 2yua_A |
99 |
66 |
57 |
1.94 |
17.54 |
76.230 |
2.800 |
T P |
| 2ys8_A |
90 |
66 |
52 |
1.42 |
26.92 |
76.130 |
3.418 |
T P |
| 2och_A |
66 |
66 |
54 |
1.98 |
31.48 |
75.074 |
2.596 |
T P |
| 1fpo_A |
171 |
66 |
56 |
1.77 |
16.07 |
74.619 |
3.002 |
T P |
| 1nz6_B |
99 |
66 |
53 |
1.76 |
11.32 |
74.615 |
2.847 |
T P |
| 2qwo_B |
92 |
66 |
53 |
1.76 |
11.32 |
73.951 |
2.850 |
T P |
| 2qwn_B |
94 |
66 |
53 |
1.75 |
11.32 |
73.832 |
2.869 |
T P |
| 1gh6_A |
114 |
66 |
50 |
1.89 |
28.00 |
69.167 |
2.512 |
T P |
| 1n4c_A |
182 |
66 |
55 |
2.09 |
14.55 |
65.624 |
2.516 |
T P |
| 2pf4_E |
163 |
66 |
51 |
2.17 |
29.41 |
65.000 |
2.243 |
T P |
| 1hdj_A |
77 |
66 |
54 |
2.08 |
27.78 |
63.653 |
2.473 |
T P |
| 1bqz_A |
77 |
66 |
55 |
2.16 |
36.36 |
62.356 |
2.429 |
T P |
| 1bq0_A |
77 |
66 |
55 |
2.35 |
34.55 |
61.784 |
2.244 |
T P |
| 1iur_A |
88 |
66 |
45 |
1.90 |
17.78 |
59.987 |
2.254 |
T P |
| 2qam_T |
93 |
66 |
28 |
2.37 |
25.00 |
34.964 |
1.132 |
T P |
| 2j28_T |
99 |
66 |
26 |
2.15 |
23.08 |
30.128 |
1.157 |
T P |
| 3fik_T |
93 |
66 |
30 |
2.53 |
3.33 |
30.084 |
1.139 |
T P |
| 2gya_R |
92 |
66 |
25 |
2.63 |
8.00 |
25.874 |
0.917 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]