LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_184.5wLII_11181_58
Total number of 3D structures: 59
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
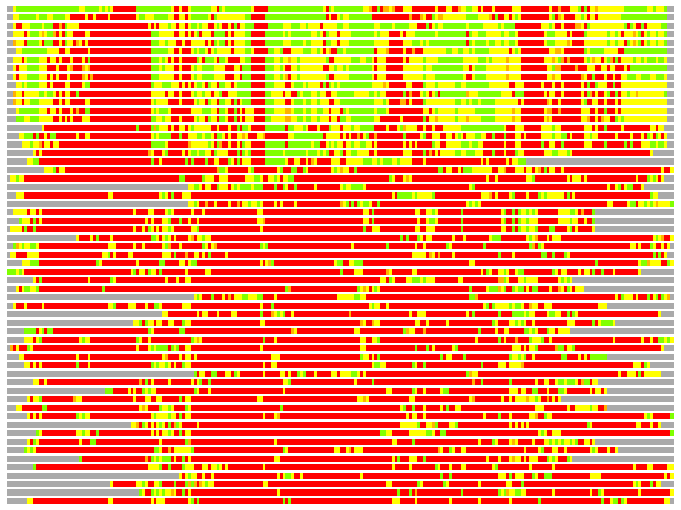
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1u0b_B |
461 |
232 |
163 |
2.11 |
12.88 |
57.900 |
7.380 |
T P |
| 3c8z_B |
401 |
232 |
157 |
2.29 |
10.83 |
51.443 |
6.561 |
T P |
| 1gax_A |
862 |
232 |
159 |
2.28 |
6.29 |
48.714 |
6.672 |
T P |
| 1wz2_A |
948 |
232 |
163 |
2.37 |
6.13 |
47.222 |
6.599 |
T P |
| 1wkb_A |
791 |
232 |
163 |
2.35 |
5.52 |
47.021 |
6.643 |
T P |
| 1ffy_A |
917 |
232 |
149 |
2.28 |
6.71 |
44.543 |
6.271 |
T P |
| 1qqt_A |
546 |
232 |
151 |
2.48 |
3.97 |
43.093 |
5.855 |
T P |
| 1f4l_A |
545 |
232 |
147 |
2.49 |
4.08 |
41.616 |
5.678 |
T P |
| 1pfv_A |
547 |
232 |
146 |
2.48 |
3.42 |
41.178 |
5.666 |
T P |
| 2ct8_A |
465 |
232 |
146 |
2.54 |
10.96 |
40.197 |
5.534 |
T P |
| 1ile_A |
821 |
232 |
143 |
2.46 |
4.90 |
39.845 |
5.586 |
T P |
| 2d54_A |
500 |
232 |
142 |
2.54 |
8.45 |
39.665 |
5.386 |
T P |
| 1a8h_A |
500 |
232 |
142 |
2.56 |
10.56 |
39.242 |
5.341 |
T P |
| 2d5b_A |
500 |
232 |
142 |
2.63 |
9.86 |
38.893 |
5.209 |
T P |
| 1f7u_A |
606 |
232 |
107 |
2.55 |
9.35 |
31.747 |
4.030 |
T P |
| 1iq0_A |
588 |
232 |
109 |
2.58 |
6.42 |
31.207 |
4.064 |
T P |
| 3fnr_A |
449 |
232 |
108 |
2.52 |
6.48 |
30.341 |
4.127 |
T P |
| 1irx_B |
508 |
232 |
95 |
2.41 |
11.58 |
29.128 |
3.786 |
T P |
| 2q1i_A |
308 |
232 |
65 |
2.38 |
7.69 |
19.313 |
2.626 |
T P |
| 1m9s_A |
523 |
232 |
56 |
2.68 |
7.14 |
15.129 |
2.014 |
T P |
| 3cvr_A |
477 |
232 |
49 |
2.72 |
6.12 |
14.114 |
1.738 |
T P |
| 3e6j_A |
219 |
232 |
45 |
2.37 |
2.22 |
13.586 |
1.825 |
T P |
| 2z66_A |
306 |
232 |
48 |
2.67 |
4.17 |
13.385 |
1.730 |
T P |
| 2z63_A |
570 |
232 |
50 |
2.77 |
8.00 |
13.298 |
1.741 |
T P |
| 2omw_A |
461 |
232 |
47 |
2.71 |
10.64 |
13.164 |
1.670 |
T P |
| 2omt_A |
461 |
232 |
45 |
2.62 |
8.89 |
13.031 |
1.652 |
T P |
| 2omy_A |
460 |
232 |
45 |
2.77 |
8.89 |
12.940 |
1.570 |
T P |
| 2z64_A |
599 |
232 |
43 |
2.69 |
4.65 |
12.841 |
1.540 |
T P |
| 1h6u_A |
308 |
232 |
43 |
2.69 |
6.98 |
12.569 |
1.541 |
T P |
| 3bz5_A |
450 |
232 |
41 |
2.72 |
2.44 |
12.451 |
1.453 |
T P |
| 1ogq_A |
313 |
232 |
41 |
2.37 |
0.00 |
12.257 |
1.663 |
T P |
| 1gwb_B |
276 |
232 |
46 |
2.74 |
4.35 |
12.219 |
1.620 |
T P |
| 1jl5_A |
353 |
232 |
41 |
2.73 |
9.76 |
12.116 |
1.447 |
T P |
| 2ft3_F |
305 |
232 |
40 |
2.59 |
5.00 |
11.931 |
1.486 |
T P |
| 1sq0_B |
265 |
232 |
42 |
2.73 |
9.52 |
11.913 |
1.484 |
T P |
| 1ziw_A |
629 |
232 |
45 |
2.71 |
6.67 |
11.850 |
1.604 |
T P |
| 1p8v_A |
277 |
232 |
40 |
2.59 |
15.00 |
11.848 |
1.488 |
T P |
| 2z62_A |
276 |
232 |
43 |
2.76 |
11.63 |
11.694 |
1.505 |
T P |
| 1xec_A |
305 |
232 |
37 |
2.49 |
5.41 |
11.693 |
1.430 |
T P |
| 1p9a_G |
266 |
232 |
41 |
2.76 |
7.32 |
11.664 |
1.433 |
T P |
| 3cig_A |
663 |
232 |
39 |
2.54 |
7.69 |
11.564 |
1.478 |
T P |
| 2id5_B |
473 |
232 |
39 |
2.71 |
7.69 |
11.472 |
1.389 |
T P |
| 1p8t_A |
285 |
232 |
42 |
2.88 |
9.52 |
11.438 |
1.411 |
T P |
| 2o6s_A |
208 |
232 |
42 |
2.88 |
7.14 |
11.385 |
1.409 |
T P |
| 2omx_A |
462 |
232 |
35 |
2.64 |
8.57 |
11.314 |
1.276 |
T P |
| 1o6v_A |
462 |
232 |
37 |
2.70 |
2.70 |
11.140 |
1.320 |
T P |
| 1u0n_D |
265 |
232 |
39 |
3.12 |
10.26 |
11.130 |
1.210 |
T P |
| 3fxi_A |
601 |
232 |
36 |
2.76 |
0.00 |
11.045 |
1.257 |
T P |
| 1m0z_B |
266 |
232 |
40 |
2.74 |
7.50 |
11.043 |
1.407 |
T P |
| 2omu_A |
462 |
232 |
38 |
2.70 |
13.16 |
10.896 |
1.356 |
T P |
| 2a0z_A |
671 |
232 |
39 |
2.66 |
0.00 |
10.871 |
1.412 |
T P |
| 2omz_A |
465 |
232 |
35 |
2.67 |
2.86 |
10.796 |
1.262 |
T P |
| 1w8a_A |
189 |
232 |
41 |
2.80 |
12.20 |
10.641 |
1.412 |
T P |
| 1ozn_A |
284 |
232 |
36 |
2.69 |
5.56 |
10.435 |
1.291 |
T P |
| 1ook_G |
282 |
232 |
35 |
2.69 |
5.71 |
10.316 |
1.255 |
T P |
| 2v70_A |
210 |
232 |
34 |
2.66 |
2.94 |
10.305 |
1.231 |
T P |
| 1xku_A |
305 |
232 |
36 |
2.75 |
5.56 |
10.029 |
1.262 |
T P |
| 2o6q_A |
270 |
232 |
34 |
2.59 |
2.94 |
9.896 |
1.266 |
T P |
| 1m10_B |
267 |
232 |
29 |
2.77 |
3.45 |
8.948 |
1.012 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]