LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_188.5wLII_11181_70
Total number of 3D structures: 56
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
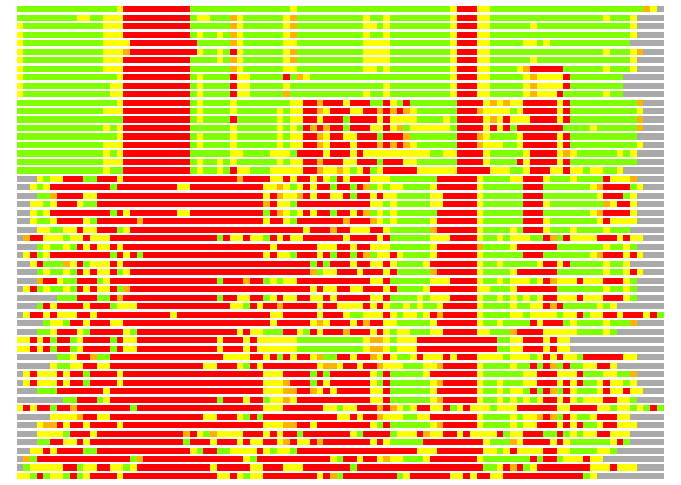
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2gj3_A |
119 |
97 |
83 |
1.11 |
20.48 |
83.709 |
6.863 |
T P |
| 2z6c_A |
121 |
97 |
80 |
1.71 |
16.25 |
75.561 |
4.428 |
T P |
| 2v1b_A |
144 |
97 |
80 |
1.65 |
12.50 |
75.457 |
4.578 |
T P |
| 1n9l_A |
109 |
97 |
80 |
1.69 |
16.25 |
75.217 |
4.467 |
T P |
| 1jnu_A |
104 |
97 |
79 |
1.79 |
12.66 |
73.784 |
4.190 |
T P |
| 2z6d_A |
118 |
97 |
80 |
1.73 |
16.25 |
69.283 |
4.368 |
T P |
| 2v0u_A |
146 |
97 |
80 |
1.66 |
12.50 |
67.254 |
4.548 |
T P |
| 2pr5_A |
127 |
97 |
75 |
1.69 |
16.00 |
66.721 |
4.196 |
T P |
| 2pd8_A |
148 |
97 |
75 |
1.82 |
17.33 |
66.694 |
3.907 |
T P |
| 3d72_A |
149 |
97 |
76 |
1.79 |
17.11 |
65.948 |
4.016 |
T P |
| 2pdr_A |
147 |
97 |
76 |
1.83 |
17.11 |
65.654 |
3.947 |
T P |
| 1bv6_A |
119 |
97 |
64 |
1.88 |
14.06 |
52.411 |
3.224 |
T P |
| 1y28_A |
119 |
97 |
66 |
2.17 |
13.64 |
51.625 |
2.913 |
T P |
| 1lsw_A |
117 |
97 |
63 |
2.01 |
12.70 |
51.424 |
2.990 |
T P |
| 1xj3_A |
116 |
97 |
63 |
2.16 |
14.29 |
51.340 |
2.791 |
T P |
| 2vv6_D |
107 |
97 |
64 |
1.94 |
14.06 |
51.267 |
3.134 |
T P |
| 2cmn_A |
117 |
97 |
65 |
2.15 |
13.85 |
51.109 |
2.894 |
T P |
| 1s67_L |
119 |
97 |
67 |
2.09 |
16.42 |
50.409 |
3.062 |
T P |
| 1d06_A |
130 |
97 |
62 |
1.79 |
11.29 |
50.369 |
3.273 |
T P |
| 1v9y_A |
103 |
97 |
63 |
2.29 |
15.87 |
47.819 |
2.640 |
T P |
| 3equ_A |
429 |
97 |
45 |
2.34 |
4.44 |
36.010 |
1.844 |
T P |
| 1k25_B |
575 |
97 |
44 |
2.37 |
11.36 |
34.755 |
1.780 |
T P |
| 3eqv_A |
443 |
97 |
42 |
2.47 |
7.14 |
33.860 |
1.633 |
T P |
| 1pmd_A |
675 |
97 |
41 |
2.10 |
7.32 |
33.834 |
1.865 |
T P |
| 1pyy_A |
607 |
97 |
43 |
2.36 |
9.30 |
33.548 |
1.751 |
T P |
| 1rp5_A |
687 |
97 |
41 |
2.36 |
7.32 |
33.539 |
1.666 |
T P |
| 2waf_A |
619 |
97 |
43 |
2.37 |
6.98 |
33.417 |
1.744 |
T P |
| 1e3u_D |
242 |
97 |
49 |
2.69 |
8.16 |
33.004 |
1.759 |
T P |
| 2c6w_B |
384 |
97 |
43 |
2.36 |
11.63 |
32.614 |
1.751 |
T P |
| 1qme_A |
558 |
97 |
41 |
2.24 |
9.76 |
32.493 |
1.749 |
T P |
| 2jc7_A |
244 |
97 |
42 |
2.26 |
7.14 |
32.310 |
1.776 |
T P |
| 2uwy_A |
469 |
97 |
41 |
2.26 |
9.76 |
31.913 |
1.737 |
T P |
| 1h8z_A |
237 |
97 |
43 |
2.52 |
9.30 |
31.660 |
1.641 |
T P |
| 2uwx_A |
468 |
97 |
41 |
2.24 |
9.76 |
31.563 |
1.749 |
T P |
| 1e3u_A |
243 |
97 |
45 |
2.54 |
11.11 |
31.448 |
1.702 |
T P |
| 1mwr_B |
629 |
97 |
46 |
2.58 |
6.52 |
31.290 |
1.718 |
T P |
| 2wad_C |
607 |
97 |
44 |
2.63 |
2.27 |
30.978 |
1.614 |
T P |
| 1k6r_A |
246 |
97 |
41 |
2.55 |
12.20 |
30.950 |
1.548 |
T P |
| 2zc3_E |
367 |
97 |
44 |
2.43 |
4.55 |
30.906 |
1.742 |
T P |
| 1mwx_A |
635 |
97 |
42 |
2.68 |
2.38 |
30.737 |
1.511 |
T P |
| 1vqq_A |
635 |
97 |
42 |
2.68 |
2.38 |
30.737 |
1.511 |
T P |
| 1nrf_A |
246 |
97 |
44 |
2.78 |
11.36 |
30.664 |
1.530 |
T P |
| 2bg1_A |
455 |
97 |
43 |
2.66 |
4.65 |
30.264 |
1.560 |
T P |
| 2rl3_B |
248 |
97 |
40 |
2.53 |
12.50 |
30.091 |
1.519 |
T P |
| 2hpb_B |
247 |
97 |
41 |
2.68 |
12.20 |
29.501 |
1.474 |
T P |
| 2iwb_A |
246 |
97 |
41 |
2.61 |
12.20 |
29.344 |
1.514 |
T P |
| 1fof_A |
246 |
97 |
40 |
2.33 |
17.50 |
29.106 |
1.643 |
T P |
| 2zc6_B |
384 |
97 |
40 |
2.98 |
7.50 |
28.622 |
1.300 |
T P |
| 2fff_B |
453 |
97 |
41 |
2.69 |
4.88 |
28.610 |
1.471 |
T P |
| 2hp5_A |
246 |
97 |
39 |
2.72 |
10.26 |
28.484 |
1.383 |
T P |
| 1mws_A |
633 |
97 |
44 |
2.90 |
6.82 |
28.468 |
1.468 |
T P |
| 2c5w_B |
385 |
97 |
38 |
2.56 |
5.26 |
27.968 |
1.430 |
T P |
| 2wae_A |
607 |
97 |
38 |
2.59 |
7.89 |
25.544 |
1.410 |
T P |
| 2jch_A |
461 |
97 |
32 |
2.56 |
6.25 |
24.080 |
1.203 |
T P |
| 2bis_A |
440 |
97 |
32 |
2.83 |
3.12 |
21.575 |
1.094 |
T P |
| 3c4s_A |
57 |
97 |
28 |
2.64 |
0.00 |
19.024 |
1.021 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]