LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_19.5wLII_10580_1
Total number of 3D structures: 71
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
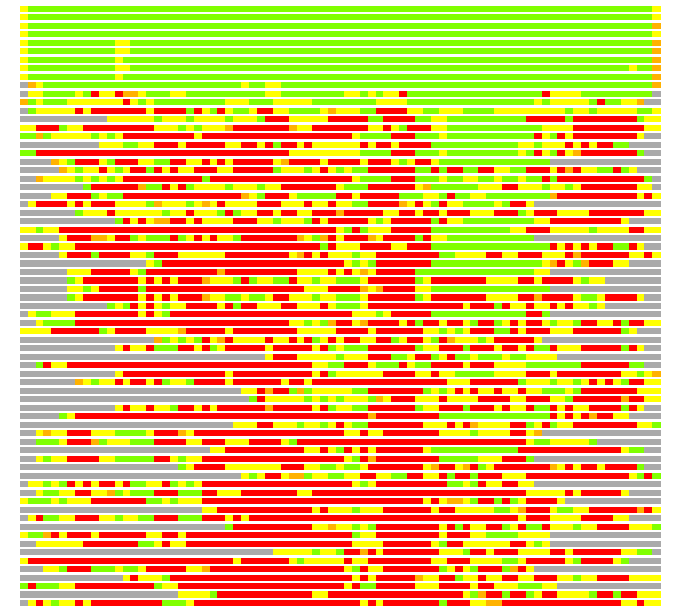
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1jk3_A |
158 |
81 |
81 |
0.69 |
17.28 |
99.172 |
10.249 |
T P |
| 1y93_A |
158 |
81 |
81 |
0.79 |
16.05 |
98.711 |
9.051 |
T P |
| 3f17_A |
158 |
81 |
81 |
0.82 |
16.05 |
98.560 |
8.813 |
T P |
| 1os9_A |
165 |
81 |
81 |
0.84 |
16.05 |
98.240 |
8.659 |
T P |
| 1utt_A |
159 |
81 |
81 |
0.96 |
16.05 |
98.191 |
7.658 |
T P |
| 2w0d_C |
159 |
81 |
81 |
0.93 |
17.28 |
98.094 |
7.893 |
T P |
| 1jiz_A |
166 |
81 |
81 |
0.96 |
16.05 |
97.986 |
7.655 |
T P |
| 3ba0_A |
365 |
81 |
81 |
1.07 |
16.05 |
97.807 |
6.909 |
T P |
| 1ros_A |
159 |
81 |
81 |
1.00 |
16.05 |
97.347 |
7.349 |
T P |
| 2poj_A |
164 |
81 |
80 |
1.20 |
17.50 |
95.609 |
6.174 |
T P |
| 2z2d_A |
158 |
81 |
75 |
1.82 |
14.67 |
85.675 |
3.908 |
T P |
| 2k2g_A |
165 |
81 |
77 |
2.22 |
15.58 |
77.956 |
3.325 |
T P |
| 2k9c_A |
152 |
81 |
60 |
2.48 |
13.33 |
48.834 |
2.329 |
T P |
| 2csd_A |
516 |
81 |
45 |
2.28 |
11.11 |
37.953 |
1.893 |
T P |
| 2uxu_A |
206 |
81 |
42 |
2.53 |
4.76 |
37.461 |
1.595 |
T P |
| 1kvs_A |
338 |
81 |
38 |
2.19 |
2.63 |
36.876 |
1.658 |
T P |
| 1t56_A |
193 |
81 |
42 |
2.52 |
7.14 |
35.588 |
1.602 |
T P |
| 1kvu_A |
338 |
81 |
35 |
2.19 |
11.43 |
35.512 |
1.528 |
T P |
| 1kvr_A |
338 |
81 |
35 |
2.13 |
8.57 |
35.053 |
1.570 |
T P |
| 1y9q_A |
178 |
81 |
42 |
2.54 |
4.76 |
34.972 |
1.593 |
T P |
| 1a9z_A |
338 |
81 |
35 |
2.17 |
2.86 |
34.447 |
1.541 |
T P |
| 2ras_B |
204 |
81 |
42 |
2.68 |
2.38 |
33.708 |
1.510 |
T P |
| 2a6h_F |
345 |
81 |
37 |
2.24 |
10.81 |
33.666 |
1.580 |
T P |
| 3fks_F |
471 |
81 |
42 |
2.71 |
2.38 |
33.253 |
1.492 |
T P |
| 2hld_D |
470 |
81 |
39 |
2.98 |
2.56 |
32.409 |
1.267 |
T P |
| 2qfc_A |
284 |
81 |
36 |
2.79 |
13.89 |
31.829 |
1.247 |
T P |
| 3dxj_F |
349 |
81 |
35 |
2.49 |
5.71 |
31.665 |
1.354 |
T P |
| 2r0q_C |
193 |
81 |
36 |
2.60 |
5.56 |
31.427 |
1.334 |
T P |
| 1u9o_A |
194 |
81 |
32 |
2.30 |
9.38 |
30.395 |
1.334 |
T P |
| 1zvv_A |
332 |
81 |
35 |
2.73 |
0.00 |
29.771 |
1.237 |
T P |
| 2a6c_A |
76 |
81 |
27 |
2.04 |
7.41 |
29.676 |
1.260 |
T P |
| 2eby_A |
102 |
81 |
31 |
2.36 |
0.00 |
29.440 |
1.262 |
T P |
| 1udb_A |
338 |
81 |
35 |
2.64 |
8.57 |
28.779 |
1.279 |
T P |
| 3cec_A |
91 |
81 |
28 |
2.16 |
7.14 |
28.436 |
1.240 |
T P |
| 1kvt_A |
338 |
81 |
36 |
2.76 |
8.33 |
28.284 |
1.257 |
T P |
| 1udc_A |
338 |
81 |
32 |
2.70 |
0.00 |
27.289 |
1.144 |
T P |
| 3clc_B |
77 |
81 |
27 |
2.06 |
3.70 |
27.086 |
1.251 |
T P |
| 3bdn_A |
234 |
81 |
31 |
2.58 |
9.68 |
26.735 |
1.157 |
T P |
| 1nr3_A |
122 |
81 |
32 |
3.01 |
3.12 |
26.552 |
1.029 |
T P |
| 3c64_A |
152 |
81 |
31 |
2.84 |
6.45 |
26.441 |
1.054 |
T P |
| 1a9y_A |
338 |
81 |
29 |
2.43 |
6.90 |
26.258 |
1.147 |
T P |
| 1ic8_B |
173 |
81 |
28 |
2.36 |
7.14 |
26.236 |
1.137 |
T P |
| 2ef8_A |
84 |
81 |
28 |
2.17 |
7.14 |
25.851 |
1.231 |
T P |
| 3dnv_B |
71 |
81 |
29 |
2.68 |
0.00 |
25.849 |
1.044 |
T P |
| 2bnm_A |
194 |
81 |
31 |
2.78 |
9.68 |
25.777 |
1.075 |
T P |
| 2b5a_A |
77 |
81 |
32 |
2.71 |
6.25 |
25.589 |
1.140 |
T P |
| 3f6w_A |
82 |
81 |
32 |
2.74 |
3.12 |
25.199 |
1.127 |
T P |
| 1kvq_A |
338 |
81 |
29 |
2.70 |
10.34 |
25.043 |
1.035 |
T P |
| 3eus_A |
85 |
81 |
22 |
1.99 |
0.00 |
24.418 |
1.053 |
T P |
| 2ewt_A |
71 |
81 |
31 |
2.72 |
0.00 |
24.341 |
1.100 |
T P |
| 2r63_A |
63 |
81 |
29 |
2.90 |
0.00 |
23.933 |
0.967 |
T P |
| 1utx_A |
66 |
81 |
28 |
2.46 |
0.00 |
23.718 |
1.092 |
T P |
| 1r63_A |
63 |
81 |
23 |
2.25 |
0.00 |
23.369 |
0.979 |
T P |
| 3bs3_A |
62 |
81 |
27 |
2.51 |
3.70 |
23.128 |
1.034 |
T P |
| 1lrk_A |
338 |
81 |
28 |
2.91 |
7.14 |
23.123 |
0.929 |
T P |
| 2ofy_A |
82 |
81 |
27 |
2.64 |
3.70 |
22.625 |
0.984 |
T P |
| 1r69_A |
63 |
81 |
27 |
2.49 |
7.41 |
22.486 |
1.041 |
T P |
| 2r1j_L |
66 |
81 |
28 |
2.62 |
0.00 |
22.410 |
1.030 |
T P |
| 1adr_A |
76 |
81 |
26 |
2.65 |
0.00 |
21.921 |
0.945 |
T P |
| 1sq8_A |
64 |
81 |
27 |
2.93 |
0.00 |
21.735 |
0.891 |
T P |
| 1b0n_A |
103 |
81 |
24 |
2.64 |
4.17 |
21.735 |
0.877 |
T P |
| 1y7y_A |
69 |
81 |
27 |
2.74 |
0.00 |
21.715 |
0.951 |
T P |
| 2k9q_A |
77 |
81 |
23 |
2.54 |
4.35 |
21.250 |
0.870 |
T P |
| 1lli_B |
92 |
81 |
26 |
2.79 |
23.08 |
21.135 |
0.901 |
T P |
| 1x57_A |
91 |
81 |
25 |
2.87 |
12.00 |
21.023 |
0.841 |
T P |
| 2csb_A |
517 |
81 |
24 |
2.87 |
0.00 |
20.711 |
0.808 |
T P |
| 2cro_A |
65 |
81 |
24 |
2.59 |
4.17 |
20.415 |
0.891 |
T P |
| 3f52_E |
78 |
81 |
26 |
2.98 |
0.00 |
20.245 |
0.845 |
T P |
| 2h8r_A |
176 |
81 |
24 |
2.58 |
20.83 |
20.061 |
0.897 |
T P |
| 3b7h_A |
76 |
81 |
22 |
2.79 |
9.09 |
18.389 |
0.762 |
T P |
| 2jvl_A |
107 |
81 |
21 |
2.92 |
0.00 |
16.808 |
0.695 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]