LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_190.5wLII_11181_77
Total number of 3D structures: 63
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
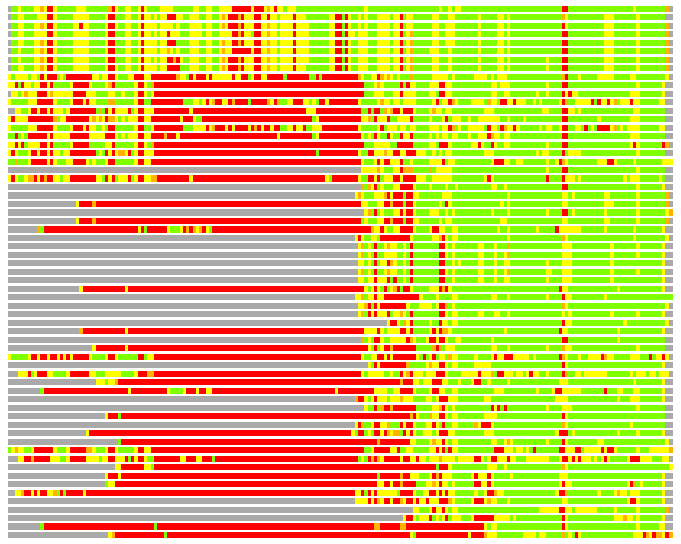
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2vqa_A |
356 |
205 |
187 |
1.68 |
20.32 |
83.661 |
10.525 |
T P |
| 2uy8_A |
377 |
205 |
182 |
2.02 |
15.38 |
66.339 |
8.596 |
T P |
| 2uy9_A |
377 |
205 |
185 |
2.08 |
16.22 |
66.029 |
8.481 |
T P |
| 2v09_A |
377 |
205 |
185 |
2.11 |
16.22 |
65.798 |
8.358 |
T P |
| 2uya_A |
375 |
205 |
183 |
2.08 |
15.85 |
65.706 |
8.406 |
T P |
| 2uyb_A |
377 |
205 |
183 |
2.07 |
15.85 |
65.674 |
8.416 |
T P |
| 1l3j_A |
372 |
205 |
182 |
2.08 |
15.93 |
65.657 |
8.345 |
T P |
| 1j58_A |
372 |
205 |
181 |
2.07 |
16.02 |
65.167 |
8.347 |
T P |
| 1uij_A |
377 |
205 |
143 |
2.23 |
5.59 |
52.642 |
6.143 |
T P |
| 1sef_A |
250 |
205 |
119 |
1.81 |
5.88 |
46.179 |
6.241 |
T P |
| 1sfn_A |
245 |
205 |
123 |
2.17 |
12.20 |
45.984 |
5.430 |
T P |
| 1ipk_C |
379 |
205 |
144 |
2.46 |
6.25 |
45.589 |
5.635 |
T P |
| 2phd_D |
352 |
205 |
113 |
2.08 |
10.62 |
44.219 |
5.182 |
T P |
| 2h0v_A |
335 |
205 |
119 |
2.27 |
14.29 |
43.985 |
5.018 |
T P |
| 1ipj_C |
368 |
205 |
141 |
2.43 |
4.96 |
43.841 |
5.570 |
T P |
| 1y3t_A |
330 |
205 |
117 |
2.22 |
12.82 |
43.072 |
5.048 |
T P |
| 1rc6_A |
242 |
205 |
112 |
1.98 |
9.82 |
42.792 |
5.390 |
T P |
| 3bu7_B |
358 |
205 |
114 |
2.17 |
11.40 |
42.488 |
5.016 |
T P |
| 2p17_A |
249 |
205 |
115 |
2.03 |
12.17 |
42.101 |
5.397 |
T P |
| 1lr5_A |
160 |
205 |
91 |
1.74 |
16.48 |
41.964 |
4.938 |
T P |
| 2d40_A |
289 |
205 |
114 |
2.31 |
7.02 |
41.873 |
4.736 |
T P |
| 1o5n_A |
114 |
205 |
87 |
1.60 |
16.09 |
40.917 |
5.120 |
T P |
| 2ete_A |
201 |
205 |
91 |
1.77 |
19.78 |
40.900 |
4.860 |
T P |
| 1fi2_A |
201 |
205 |
89 |
1.68 |
20.22 |
40.899 |
4.999 |
T P |
| 1o4t_A |
115 |
205 |
89 |
1.64 |
15.73 |
40.785 |
5.128 |
T P |
| 2et7_A |
201 |
205 |
90 |
1.75 |
21.11 |
40.679 |
4.866 |
T P |
| 2q1z_B |
185 |
205 |
98 |
2.06 |
20.41 |
39.767 |
4.529 |
T P |
| 2b8m_A |
109 |
205 |
86 |
1.61 |
10.47 |
39.709 |
5.024 |
T P |
| 1x7n_A |
189 |
205 |
91 |
1.80 |
15.38 |
39.694 |
4.796 |
T P |
| 1plz_A |
187 |
205 |
91 |
1.81 |
15.38 |
39.392 |
4.772 |
T P |
| 1qxr_A |
187 |
205 |
90 |
1.77 |
15.56 |
39.373 |
4.819 |
T P |
| 1x82_A |
190 |
205 |
91 |
1.82 |
15.38 |
39.271 |
4.748 |
T P |
| 1j3q_A |
187 |
205 |
89 |
1.73 |
15.73 |
39.107 |
4.868 |
T P |
| 3bal_A |
149 |
205 |
87 |
1.73 |
18.39 |
39.068 |
4.749 |
T P |
| 3cew_A |
118 |
205 |
85 |
1.58 |
10.59 |
39.054 |
5.049 |
T P |
| 1yhf_A |
114 |
205 |
84 |
1.69 |
11.90 |
38.922 |
4.703 |
T P |
| 2gc1_A |
188 |
205 |
89 |
1.83 |
14.61 |
38.744 |
4.609 |
T P |
| 2f4p_A |
134 |
205 |
82 |
1.53 |
18.29 |
38.551 |
5.045 |
T P |
| 2o1q_A |
144 |
205 |
88 |
1.90 |
14.77 |
38.385 |
4.400 |
T P |
| 2pfw_A |
112 |
205 |
85 |
1.69 |
14.12 |
38.179 |
4.760 |
T P |
| 2ilb_A |
114 |
205 |
83 |
1.77 |
15.66 |
37.970 |
4.427 |
T P |
| 1tq5_A |
234 |
205 |
103 |
2.30 |
3.88 |
36.912 |
4.288 |
T P |
| 2ozj_A |
110 |
205 |
80 |
1.70 |
16.25 |
36.902 |
4.449 |
T P |
| 1sq4_A |
270 |
205 |
114 |
2.44 |
12.28 |
36.363 |
4.489 |
T P |
| 3d82_A |
102 |
205 |
81 |
1.82 |
20.99 |
36.190 |
4.216 |
T P |
| 2z2s_B |
182 |
205 |
93 |
2.13 |
21.51 |
36.146 |
4.178 |
T P |
| 3ebr_A |
157 |
205 |
90 |
2.11 |
14.44 |
36.099 |
4.069 |
T P |
| 2gu9_A |
111 |
205 |
79 |
1.73 |
12.66 |
35.958 |
4.318 |
T P |
| 3fjs_C |
107 |
205 |
77 |
1.58 |
18.18 |
35.871 |
4.590 |
T P |
| 2bnm_A |
194 |
205 |
85 |
1.79 |
14.12 |
35.820 |
4.488 |
T P |
| 3cjx_A |
162 |
205 |
85 |
1.94 |
15.29 |
35.414 |
4.170 |
T P |
| 2q30_A |
106 |
205 |
80 |
1.88 |
20.00 |
34.912 |
4.043 |
T P |
| 1j1l_A |
288 |
205 |
104 |
2.29 |
6.73 |
34.790 |
4.344 |
T P |
| 2qnk_A |
286 |
205 |
102 |
2.66 |
8.82 |
34.591 |
3.699 |
T P |
| 2yu2_A |
387 |
205 |
75 |
1.57 |
10.67 |
34.165 |
4.487 |
T P |
| 2i45_D |
99 |
205 |
79 |
1.87 |
18.99 |
34.128 |
4.005 |
T P |
| 1yfu_A |
174 |
205 |
78 |
1.95 |
12.82 |
33.296 |
3.811 |
T P |
| 2qdr_A |
285 |
205 |
92 |
2.33 |
16.30 |
32.686 |
3.787 |
T P |
| 1zvf_A |
176 |
205 |
78 |
2.12 |
12.82 |
31.755 |
3.508 |
T P |
| 2fqp_A |
95 |
205 |
75 |
1.94 |
13.33 |
28.378 |
3.685 |
T P |
| 2o8q_A |
122 |
205 |
70 |
1.95 |
20.00 |
27.317 |
3.415 |
T P |
| 1vr3_A |
179 |
205 |
61 |
1.86 |
19.67 |
26.973 |
3.117 |
T P |
| 1m4o_A |
179 |
205 |
60 |
2.42 |
13.33 |
23.539 |
2.384 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]