LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_191.5wLII_11181_78
Total number of 3D structures: 25
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
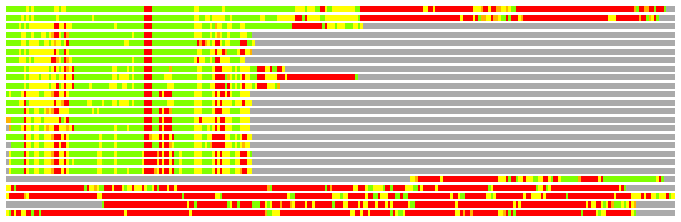
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1sr9_B |
577 |
266 |
158 |
1.97 |
22.78 |
52.915 |
7.625 |
T P |
| 3fig_B |
577 |
266 |
157 |
2.18 |
22.29 |
50.375 |
6.890 |
T P |
| 3ble_A |
307 |
266 |
123 |
1.87 |
14.63 |
41.020 |
6.239 |
T P |
| 3eeg_B |
273 |
266 |
92 |
1.81 |
15.22 |
31.346 |
4.806 |
T P |
| 3dxi_A |
299 |
266 |
88 |
1.77 |
14.77 |
30.547 |
4.716 |
T P |
| 1nvm_A |
340 |
266 |
89 |
1.87 |
16.85 |
29.990 |
4.513 |
T P |
| 3ewb_X |
271 |
266 |
86 |
1.81 |
17.44 |
29.696 |
4.499 |
T P |
| 2cw6_A |
296 |
266 |
96 |
2.01 |
13.54 |
29.568 |
4.549 |
T P |
| 2ftp_A |
300 |
266 |
97 |
2.05 |
16.49 |
29.087 |
4.504 |
T P |
| 1ydo_A |
298 |
266 |
93 |
2.08 |
16.13 |
28.166 |
4.274 |
T P |
| 3bg9_A |
601 |
266 |
82 |
1.87 |
8.54 |
26.252 |
4.153 |
T P |
| 1ydn_A |
283 |
266 |
88 |
2.18 |
19.32 |
25.858 |
3.862 |
T P |
| 2nx9_B |
453 |
266 |
85 |
2.09 |
12.94 |
25.217 |
3.878 |
T P |
| 2qf7_A |
1075 |
266 |
83 |
2.02 |
8.43 |
25.028 |
3.918 |
T P |
| 3bg3_A |
679 |
266 |
82 |
2.10 |
9.76 |
23.944 |
3.730 |
T P |
| 1rqe_A |
472 |
266 |
80 |
2.11 |
13.75 |
23.718 |
3.617 |
T P |
| 1rr2_A |
471 |
266 |
79 |
2.10 |
10.13 |
23.527 |
3.594 |
T P |
| 1rqb_A |
471 |
266 |
79 |
2.12 |
13.92 |
23.485 |
3.562 |
T P |
| 1s3h_A |
471 |
266 |
79 |
2.15 |
13.92 |
23.077 |
3.514 |
T P |
| 1u5j_A |
470 |
266 |
80 |
2.21 |
13.75 |
23.043 |
3.469 |
T P |
| 1t4o_B |
88 |
266 |
53 |
2.07 |
18.87 |
15.358 |
2.439 |
T P |
| 3bg5_A |
1137 |
266 |
56 |
2.62 |
3.57 |
14.008 |
2.056 |
T P |
| 2hqa_A |
910 |
266 |
57 |
2.70 |
1.75 |
13.692 |
2.037 |
T P |
| 2jh3_A |
459 |
266 |
47 |
2.76 |
8.51 |
11.986 |
1.646 |
T P |
| 2hnh_A |
910 |
266 |
40 |
2.62 |
7.50 |
9.927 |
1.472 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]