LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_194.5wLII_11184_3
Total number of 3D structures: 43
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
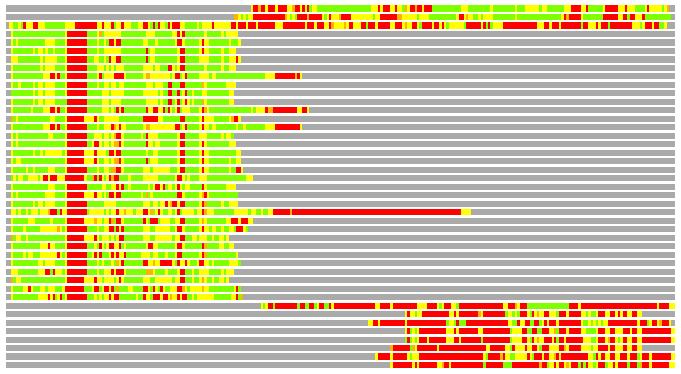
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3ezu_A |
336 |
273 |
132 |
2.19 |
14.39 |
36.164 |
5.769 |
T P |
| 1w25_A |
454 |
273 |
126 |
2.32 |
18.25 |
35.459 |
5.208 |
T P |
| 3bre_B |
328 |
273 |
135 |
2.74 |
9.63 |
30.485 |
4.759 |
T P |
| 2zay_A |
123 |
273 |
83 |
1.83 |
14.46 |
27.332 |
4.297 |
T P |
| 1xhe_B |
122 |
273 |
79 |
1.68 |
6.33 |
26.775 |
4.445 |
T P |
| 1zes_A |
121 |
273 |
80 |
1.90 |
10.00 |
25.656 |
4.000 |
T P |
| 2eub_A |
121 |
273 |
79 |
1.92 |
7.59 |
25.448 |
3.912 |
T P |
| 1b00_A |
122 |
273 |
80 |
2.01 |
10.00 |
25.400 |
3.794 |
T P |
| 1l5z_A |
146 |
273 |
91 |
2.15 |
7.69 |
25.365 |
4.049 |
T P |
| 2jb9_B |
122 |
273 |
80 |
2.04 |
8.75 |
25.171 |
3.730 |
T P |
| 2jba_B |
121 |
273 |
79 |
2.01 |
8.86 |
25.116 |
3.740 |
T P |
| 2jba_A |
125 |
273 |
79 |
2.02 |
8.86 |
24.994 |
3.726 |
T P |
| 1yio_A |
198 |
273 |
89 |
2.12 |
2.25 |
24.953 |
4.016 |
T P |
| 1mb3_A |
117 |
273 |
75 |
1.95 |
2.67 |
24.480 |
3.656 |
T P |
| 1qkk_A |
139 |
273 |
92 |
2.17 |
7.61 |
24.253 |
4.050 |
T P |
| 2a9o_A |
117 |
273 |
78 |
1.93 |
7.69 |
24.107 |
3.839 |
T P |
| 2a9r_A |
117 |
273 |
77 |
1.93 |
6.49 |
23.779 |
3.793 |
T P |
| 1zgz_A |
121 |
273 |
79 |
1.99 |
10.13 |
22.742 |
3.781 |
T P |
| 1zh2_A |
120 |
273 |
81 |
2.03 |
7.41 |
22.593 |
3.805 |
T P |
| 1ys7_B |
226 |
273 |
80 |
2.03 |
6.25 |
22.583 |
3.765 |
T P |
| 3crn_A |
128 |
273 |
79 |
2.08 |
2.53 |
22.408 |
3.620 |
T P |
| 1mvo_A |
121 |
273 |
77 |
2.01 |
5.19 |
22.109 |
3.653 |
T P |
| 1xhf_B |
122 |
273 |
78 |
1.93 |
5.13 |
22.052 |
3.848 |
T P |
| 2iyn_B |
124 |
273 |
79 |
2.15 |
8.86 |
21.856 |
3.516 |
T P |
| 1ny5_B |
385 |
273 |
94 |
2.60 |
5.32 |
21.844 |
3.482 |
T P |
| 2gwr_A |
225 |
273 |
77 |
2.17 |
6.49 |
21.681 |
3.391 |
T P |
| 1dbw_B |
125 |
273 |
79 |
2.14 |
6.33 |
21.347 |
3.533 |
T P |
| 1tmy_A |
118 |
273 |
77 |
2.23 |
3.90 |
20.986 |
3.311 |
T P |
| 2qsj_B |
122 |
273 |
73 |
2.04 |
8.22 |
20.645 |
3.410 |
T P |
| 1d5w_A |
122 |
273 |
76 |
2.17 |
5.26 |
20.501 |
3.352 |
T P |
| 1kgs_A |
219 |
273 |
74 |
2.18 |
8.11 |
20.466 |
3.253 |
T P |
| 1p2f_A |
217 |
273 |
74 |
2.16 |
8.11 |
20.420 |
3.278 |
T P |
| 1u0s_Y |
118 |
273 |
78 |
2.25 |
3.85 |
20.329 |
3.323 |
T P |
| 1dc8_A |
123 |
273 |
73 |
2.27 |
5.48 |
18.361 |
3.075 |
T P |
| 1dc7_A |
124 |
273 |
71 |
2.16 |
4.23 |
18.207 |
3.139 |
T P |
| 2oqr_A |
226 |
273 |
50 |
2.23 |
14.00 |
13.908 |
2.150 |
T P |
| 2cmn_A |
117 |
273 |
43 |
2.73 |
6.98 |
9.958 |
1.520 |
T P |
| 2pr5_A |
127 |
273 |
40 |
2.57 |
2.50 |
9.843 |
1.498 |
T P |
| 1lsw_A |
117 |
273 |
41 |
2.67 |
9.76 |
9.812 |
1.482 |
T P |
| 1xj3_A |
116 |
273 |
41 |
2.72 |
7.32 |
9.513 |
1.451 |
T P |
| 1y28_A |
119 |
273 |
39 |
2.97 |
10.26 |
9.425 |
1.270 |
T P |
| 1bv6_A |
119 |
273 |
39 |
2.78 |
2.56 |
9.000 |
1.353 |
T P |
| 2vv6_D |
107 |
273 |
36 |
2.81 |
5.56 |
8.602 |
1.236 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]