LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_197.5wLII_11184_13
Total number of 3D structures: 74
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
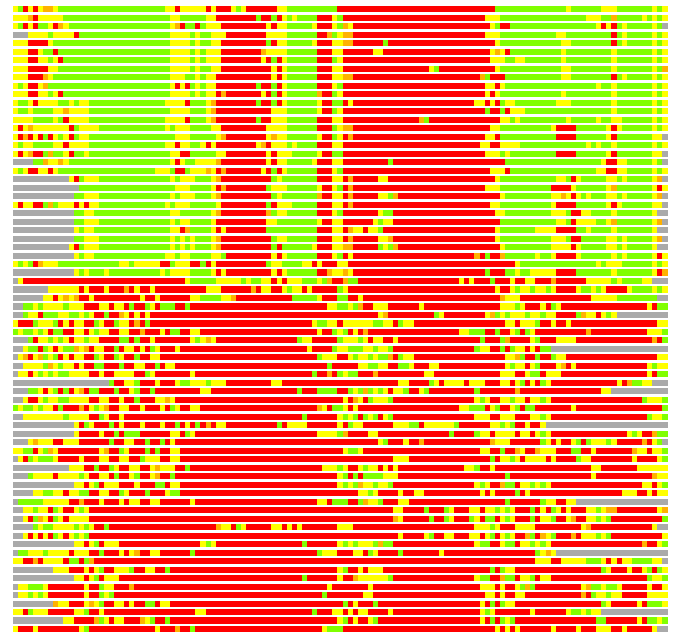
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1r62_A |
136 |
129 |
85 |
1.86 |
16.47 |
61.100 |
4.331 |
T P |
| 3d36_B |
221 |
129 |
86 |
1.86 |
6.98 |
56.790 |
4.384 |
T P |
| 3cgz_A |
143 |
129 |
79 |
1.93 |
6.33 |
53.565 |
3.889 |
T P |
| 1gkz_A |
316 |
129 |
88 |
2.06 |
4.55 |
52.571 |
4.081 |
T P |
| 2e0a_A |
358 |
129 |
84 |
1.87 |
9.52 |
52.238 |
4.271 |
T P |
| 1y8o_A |
374 |
129 |
87 |
1.92 |
8.05 |
52.162 |
4.303 |
T P |
| 2btz_A |
357 |
129 |
84 |
1.83 |
9.52 |
51.748 |
4.342 |
T P |
| 2zkj_B |
359 |
129 |
83 |
1.81 |
7.23 |
51.399 |
4.339 |
T P |
| 2q8g_A |
368 |
129 |
82 |
1.89 |
6.10 |
51.282 |
4.111 |
T P |
| 2c2a_A |
240 |
129 |
82 |
1.76 |
7.32 |
50.990 |
4.416 |
T P |
| 3crk_A |
370 |
129 |
85 |
1.89 |
9.41 |
50.240 |
4.280 |
T P |
| 1i58_A |
189 |
129 |
87 |
1.99 |
5.75 |
49.713 |
4.161 |
T P |
| 1b3q_B |
370 |
129 |
85 |
2.02 |
4.71 |
48.600 |
4.002 |
T P |
| 2ch4_A |
320 |
129 |
85 |
2.07 |
4.71 |
48.327 |
3.916 |
T P |
| 1til_A |
141 |
129 |
83 |
2.21 |
4.82 |
48.067 |
3.589 |
T P |
| 1l0o_A |
141 |
129 |
80 |
1.99 |
5.00 |
47.700 |
3.823 |
T P |
| 1i5d_A |
190 |
129 |
85 |
2.18 |
4.71 |
47.626 |
3.726 |
T P |
| 1tid_A |
136 |
129 |
78 |
1.93 |
5.13 |
47.271 |
3.847 |
T P |
| 1id0_A |
146 |
129 |
77 |
1.81 |
5.19 |
46.796 |
4.035 |
T P |
| 1ysr_A |
137 |
129 |
79 |
2.09 |
11.39 |
46.083 |
3.608 |
T P |
| 1z5b_A |
466 |
129 |
75 |
2.00 |
5.33 |
45.422 |
3.566 |
T P |
| 1pvg_A |
378 |
129 |
73 |
1.89 |
4.11 |
45.290 |
3.668 |
T P |
| 1z59_A |
462 |
129 |
77 |
2.12 |
5.19 |
45.254 |
3.463 |
T P |
| 1th8_A |
132 |
129 |
77 |
2.02 |
6.49 |
45.001 |
3.627 |
T P |
| 2q2e_B |
581 |
129 |
74 |
1.91 |
9.46 |
44.726 |
3.682 |
T P |
| 2zbk_B |
506 |
129 |
76 |
1.95 |
9.21 |
44.623 |
3.700 |
T P |
| 1ei1_A |
391 |
129 |
74 |
1.93 |
5.41 |
44.591 |
3.648 |
T P |
| 1mx0_C |
466 |
129 |
75 |
2.00 |
5.33 |
44.382 |
3.573 |
T P |
| 1mu5_A |
460 |
129 |
75 |
2.08 |
5.33 |
43.660 |
3.446 |
T P |
| 1kij_A |
384 |
129 |
71 |
1.95 |
12.68 |
43.490 |
3.459 |
T P |
| 1bxd_A |
161 |
129 |
76 |
2.19 |
9.21 |
42.005 |
3.325 |
T P |
| 1aj6_A |
194 |
129 |
70 |
2.03 |
5.71 |
41.575 |
3.282 |
T P |
| 2c11_A |
670 |
129 |
51 |
2.48 |
9.80 |
27.155 |
1.980 |
T P |
| 2cw7_A |
537 |
129 |
48 |
2.71 |
4.17 |
25.196 |
1.708 |
T P |
| 2cw8_A |
537 |
129 |
44 |
2.60 |
6.82 |
24.062 |
1.631 |
T P |
| 1v4y_A |
474 |
129 |
45 |
2.69 |
6.67 |
23.148 |
1.614 |
T P |
| 1ejx_C |
555 |
129 |
43 |
2.63 |
2.33 |
23.000 |
1.578 |
T P |
| 2gok_A |
404 |
129 |
44 |
2.63 |
4.55 |
22.977 |
1.614 |
T P |
| 1fwg_C |
565 |
129 |
44 |
2.59 |
4.55 |
22.528 |
1.634 |
T P |
| 3feq_C |
406 |
129 |
44 |
2.62 |
0.00 |
22.427 |
1.616 |
T P |
| 2vun_A |
385 |
129 |
43 |
2.65 |
2.33 |
21.573 |
1.561 |
T P |
| 2q09_A |
402 |
129 |
43 |
2.68 |
4.65 |
21.298 |
1.546 |
T P |
| 2g3f_A |
414 |
129 |
44 |
2.70 |
6.82 |
21.176 |
1.569 |
T P |
| 1rjq_A |
474 |
129 |
42 |
2.92 |
9.52 |
21.069 |
1.389 |
T P |
| 1nfg_A |
456 |
129 |
41 |
2.85 |
2.44 |
20.944 |
1.389 |
T P |
| 1k1d_A |
459 |
129 |
38 |
2.40 |
10.53 |
20.807 |
1.521 |
T P |
| 1fwh_C |
565 |
129 |
44 |
2.75 |
2.27 |
20.771 |
1.545 |
T P |
| 1ejs_C |
565 |
129 |
41 |
2.66 |
4.88 |
20.742 |
1.487 |
T P |
| 2kau_C |
565 |
129 |
40 |
2.48 |
5.00 |
20.594 |
1.551 |
T P |
| 2jmz_A |
186 |
129 |
41 |
2.70 |
4.88 |
20.228 |
1.463 |
T P |
| 1fwi_C |
550 |
129 |
37 |
2.73 |
10.81 |
20.047 |
1.306 |
T P |
| 1k75_A |
431 |
129 |
36 |
2.90 |
8.33 |
19.951 |
1.199 |
T P |
| 1gkr_A |
450 |
129 |
38 |
2.78 |
5.26 |
19.316 |
1.320 |
T P |
| 2puz_A |
404 |
129 |
37 |
2.78 |
8.11 |
18.851 |
1.283 |
T P |
| 1a5l_C |
536 |
129 |
37 |
2.94 |
2.70 |
18.775 |
1.216 |
T P |
| 1gkp_A |
457 |
129 |
34 |
2.62 |
8.82 |
18.545 |
1.252 |
T P |
| 1ejt_C |
565 |
129 |
39 |
2.75 |
7.69 |
18.475 |
1.368 |
T P |
| 1kae_A |
434 |
129 |
35 |
2.71 |
11.43 |
18.384 |
1.246 |
T P |
| 1m7j_A |
474 |
129 |
36 |
2.68 |
0.00 |
18.246 |
1.296 |
T P |
| 1eju_C |
552 |
129 |
36 |
2.92 |
16.67 |
18.181 |
1.193 |
T P |
| 1ejv_C |
552 |
129 |
34 |
2.75 |
8.82 |
18.053 |
1.192 |
T P |
| 1am2_A |
181 |
129 |
37 |
2.72 |
10.81 |
17.997 |
1.313 |
T P |
| 1fwf_C |
550 |
129 |
34 |
2.88 |
11.76 |
17.989 |
1.139 |
T P |
| 1ejr_C |
552 |
129 |
37 |
2.73 |
5.41 |
17.952 |
1.308 |
T P |
| 1rk6_A |
474 |
129 |
34 |
2.90 |
0.00 |
17.812 |
1.133 |
T P |
| 1us1_A |
704 |
129 |
32 |
2.71 |
6.25 |
17.693 |
1.139 |
T P |
| 1ef2_A |
565 |
129 |
30 |
2.73 |
3.33 |
16.167 |
1.060 |
T P |
| 1fwa_C |
565 |
129 |
32 |
2.68 |
6.25 |
16.143 |
1.153 |
T P |
| 1krc_C |
566 |
129 |
31 |
2.75 |
6.45 |
15.903 |
1.088 |
T P |
| 1krb_C |
565 |
129 |
29 |
2.83 |
3.45 |
15.742 |
0.989 |
T P |
| 1a5k_C |
566 |
129 |
29 |
2.85 |
10.34 |
15.657 |
0.985 |
T P |
| 3e74_B |
432 |
129 |
30 |
2.85 |
3.33 |
15.291 |
1.016 |
T P |
| 1a5m_C |
566 |
129 |
28 |
2.73 |
3.57 |
15.164 |
0.989 |
T P |
| 1at0_A |
145 |
129 |
21 |
2.92 |
4.76 |
11.945 |
0.695 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]