LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_202.5wLII_11184_18
Total number of 3D structures: 68
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
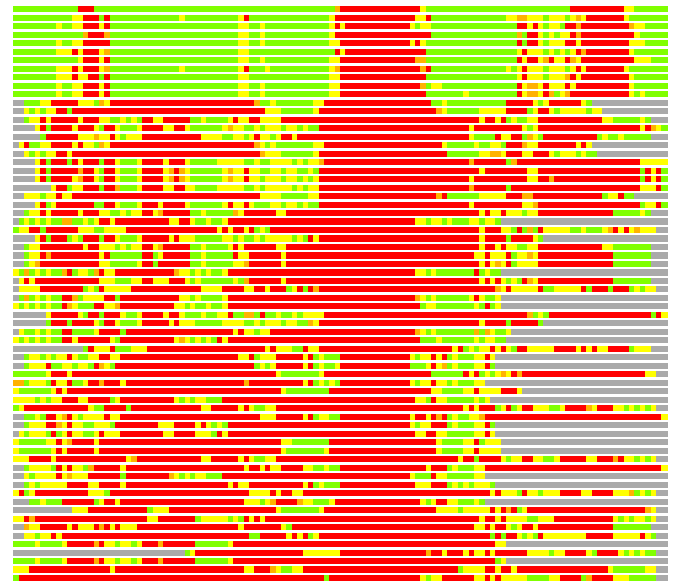
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2co7_B |
197 |
122 |
94 |
1.10 |
17.02 |
75.546 |
7.858 |
T P |
| 1l4i_A |
189 |
122 |
94 |
1.70 |
15.96 |
72.656 |
5.215 |
T P |
| 3dpa_A |
218 |
122 |
91 |
1.68 |
13.19 |
70.416 |
5.123 |
T P |
| 1qpx_A |
206 |
122 |
88 |
1.65 |
13.64 |
68.317 |
5.036 |
T P |
| 1qpp_B |
212 |
122 |
89 |
1.62 |
14.61 |
68.092 |
5.188 |
T P |
| 1klf_A |
205 |
122 |
92 |
1.69 |
13.04 |
67.583 |
5.152 |
T P |
| 2w07_A |
215 |
122 |
89 |
1.79 |
13.48 |
66.007 |
4.721 |
T P |
| 3bwu_C |
200 |
122 |
92 |
1.65 |
13.04 |
65.582 |
5.267 |
T P |
| 1qun_A |
200 |
122 |
91 |
1.69 |
12.09 |
65.288 |
5.079 |
T P |
| 2j2z_A |
216 |
122 |
90 |
1.74 |
13.33 |
65.130 |
4.892 |
T P |
| 1n0l_A |
215 |
122 |
89 |
1.83 |
13.48 |
63.801 |
4.600 |
T P |
| 2gbc_A |
730 |
122 |
43 |
2.17 |
4.65 |
27.761 |
1.897 |
T P |
| 2z3z_A |
651 |
122 |
42 |
2.38 |
2.38 |
25.358 |
1.690 |
T P |
| 1orv_A |
728 |
122 |
46 |
2.42 |
13.04 |
25.162 |
1.824 |
T P |
| 1vz3_A |
710 |
122 |
45 |
2.51 |
6.67 |
25.128 |
1.727 |
T P |
| 1z68_A |
719 |
122 |
48 |
2.57 |
4.17 |
24.931 |
1.800 |
T P |
| 2jid_A |
728 |
122 |
41 |
2.42 |
4.88 |
24.920 |
1.626 |
T P |
| 2d5l_A |
665 |
122 |
40 |
2.47 |
2.50 |
24.840 |
1.559 |
T P |
| 1o6f_A |
710 |
122 |
45 |
2.55 |
8.89 |
24.685 |
1.698 |
T P |
| 1vz2_A |
710 |
122 |
43 |
2.55 |
9.30 |
24.275 |
1.622 |
T P |
| 1h2x_A |
710 |
122 |
45 |
2.58 |
8.89 |
24.269 |
1.677 |
T P |
| 1yr2_A |
680 |
122 |
43 |
2.52 |
13.95 |
24.257 |
1.642 |
T P |
| 2dcm_A |
662 |
122 |
38 |
2.60 |
2.63 |
24.186 |
1.407 |
T P |
| 1o6g_A |
710 |
122 |
42 |
2.39 |
9.52 |
24.157 |
1.685 |
T P |
| 2bgr_A |
729 |
122 |
43 |
2.48 |
11.63 |
23.987 |
1.667 |
T P |
| 3f0d_D |
157 |
122 |
41 |
2.53 |
2.44 |
23.907 |
1.560 |
T P |
| 2qzp_B |
561 |
122 |
45 |
2.74 |
2.22 |
23.820 |
1.585 |
T P |
| 1e5t_A |
710 |
122 |
41 |
2.36 |
12.20 |
23.727 |
1.665 |
T P |
| 2g63_A |
726 |
122 |
42 |
2.52 |
11.90 |
23.630 |
1.603 |
T P |
| 1wcy_A |
729 |
122 |
41 |
2.37 |
14.63 |
23.624 |
1.658 |
T P |
| 3d4l_A |
728 |
122 |
40 |
2.34 |
15.00 |
23.478 |
1.637 |
T P |
| 2pmp_A |
160 |
122 |
42 |
2.36 |
9.52 |
23.446 |
1.707 |
T P |
| 2onc_B |
728 |
122 |
41 |
2.43 |
12.20 |
23.384 |
1.624 |
T P |
| 2qr5_A |
576 |
122 |
44 |
2.71 |
6.82 |
23.370 |
1.568 |
T P |
| 2uzh_C |
155 |
122 |
39 |
2.35 |
2.56 |
23.130 |
1.593 |
T P |
| 1knj_A |
156 |
122 |
41 |
2.25 |
7.32 |
23.114 |
1.748 |
T P |
| 3ddu_A |
707 |
122 |
40 |
2.49 |
7.50 |
22.825 |
1.547 |
T P |
| 1qfm_A |
710 |
122 |
39 |
2.40 |
10.26 |
22.670 |
1.561 |
T P |
| 1w55_A |
369 |
122 |
38 |
2.42 |
7.89 |
22.269 |
1.509 |
T P |
| 3b6n_A |
150 |
122 |
37 |
2.21 |
0.00 |
21.451 |
1.602 |
T P |
| 1r9m_B |
733 |
122 |
40 |
2.77 |
7.50 |
21.340 |
1.394 |
T P |
| 1jn1_A |
157 |
122 |
40 |
2.58 |
5.00 |
21.152 |
1.494 |
T P |
| 1vh8_A |
153 |
122 |
39 |
2.47 |
2.56 |
20.894 |
1.519 |
T P |
| 1xfd_A |
723 |
122 |
33 |
2.23 |
6.06 |
20.834 |
1.416 |
T P |
| 1h47_B |
158 |
122 |
34 |
2.64 |
2.94 |
20.527 |
1.243 |
T P |
| 2rip_A |
729 |
122 |
31 |
2.03 |
0.00 |
20.119 |
1.453 |
T P |
| 1yqn_A |
158 |
122 |
35 |
2.47 |
2.86 |
20.020 |
1.363 |
T P |
| 2qjr_A |
728 |
122 |
36 |
2.74 |
5.56 |
19.788 |
1.266 |
T P |
| 2amt_A |
156 |
122 |
34 |
2.87 |
2.94 |
19.623 |
1.146 |
T P |
| 3fpi_A |
161 |
122 |
34 |
2.45 |
5.88 |
19.293 |
1.335 |
T P |
| 2gzl_A |
159 |
122 |
35 |
2.68 |
8.57 |
19.105 |
1.259 |
T P |
| 2qt9_A |
728 |
122 |
31 |
2.23 |
0.00 |
19.068 |
1.328 |
T P |
| 1pfq_A |
728 |
122 |
30 |
2.11 |
0.00 |
19.010 |
1.359 |
T P |
| 1r9n_B |
731 |
122 |
34 |
2.78 |
5.88 |
18.699 |
1.179 |
T P |
| 1gx1_A |
157 |
122 |
32 |
2.71 |
3.12 |
18.643 |
1.139 |
T P |
| 1iv3_A |
150 |
122 |
35 |
2.58 |
5.71 |
18.585 |
1.305 |
T P |
| 1t0a_A |
158 |
122 |
32 |
2.60 |
6.25 |
18.544 |
1.185 |
T P |
| 2rgu_A |
728 |
122 |
35 |
2.68 |
5.71 |
18.349 |
1.259 |
T P |
| 1iv1_A |
150 |
122 |
32 |
2.59 |
3.12 |
18.314 |
1.189 |
T P |
| 1h2w_A |
710 |
122 |
32 |
2.70 |
0.00 |
18.213 |
1.141 |
T P |
| 1e8m_A |
710 |
122 |
35 |
2.84 |
2.86 |
17.982 |
1.190 |
T P |
| 1to7_B |
728 |
122 |
29 |
2.78 |
3.45 |
17.038 |
1.008 |
T P |
| 1va4_A |
271 |
122 |
33 |
2.81 |
3.03 |
16.949 |
1.135 |
T P |
| 2hu5_B |
575 |
122 |
28 |
2.32 |
10.71 |
16.768 |
1.159 |
T P |
| 3ccb_B |
729 |
122 |
31 |
2.89 |
6.45 |
16.739 |
1.037 |
T P |
| 2hu8_A |
575 |
122 |
28 |
2.36 |
10.71 |
16.731 |
1.138 |
T P |
| 1tk3_B |
728 |
122 |
28 |
2.63 |
10.71 |
16.557 |
1.025 |
T P |
| 2ecf_A |
700 |
122 |
30 |
2.51 |
10.00 |
16.177 |
1.150 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]