LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_203.5wLII_11184_19
Total number of 3D structures: 40
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
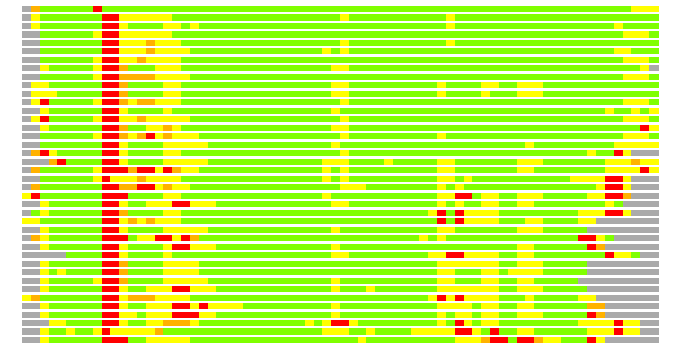
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2vi7_C |
165 |
72 |
70 |
0.97 |
21.43 |
95.797 |
6.558 |
T P |
| 2i79_A |
171 |
72 |
69 |
1.42 |
14.49 |
90.856 |
4.536 |
T P |
| 2cnt_A |
151 |
72 |
69 |
1.42 |
14.49 |
90.162 |
4.532 |
T P |
| 1yvo_B |
171 |
72 |
68 |
1.43 |
14.71 |
89.833 |
4.459 |
T P |
| 2ge3_A |
164 |
72 |
68 |
1.49 |
11.76 |
89.663 |
4.278 |
T P |
| 1vhs_A |
165 |
72 |
68 |
1.62 |
14.71 |
89.126 |
3.959 |
T P |
| 2j8m_A |
171 |
72 |
68 |
1.52 |
14.71 |
88.907 |
4.192 |
T P |
| 1y9k_B |
154 |
72 |
67 |
1.43 |
14.93 |
88.798 |
4.393 |
T P |
| 2bl1_A |
170 |
72 |
68 |
1.70 |
14.71 |
88.639 |
3.768 |
T P |
| 1j4j_A |
170 |
72 |
69 |
1.53 |
14.49 |
88.560 |
4.222 |
T P |
| 1ghe_B |
171 |
72 |
69 |
1.55 |
14.49 |
88.438 |
4.181 |
T P |
| 2jlm_D |
182 |
72 |
68 |
1.61 |
8.82 |
88.274 |
3.974 |
T P |
| 2ob0_C |
166 |
72 |
68 |
1.50 |
10.29 |
88.030 |
4.256 |
T P |
| 1yr0_A |
163 |
72 |
68 |
1.65 |
17.65 |
88.006 |
3.879 |
T P |
| 1yvk_A |
152 |
72 |
67 |
1.60 |
8.96 |
87.281 |
3.951 |
T P |
| 3dr6_A |
172 |
72 |
67 |
1.74 |
11.94 |
86.962 |
3.637 |
T P |
| 1tiq_A |
173 |
72 |
68 |
1.75 |
13.24 |
86.257 |
3.684 |
T P |
| 1wwz_A |
157 |
72 |
64 |
1.54 |
17.19 |
84.156 |
3.905 |
T P |
| 2cy2_A |
174 |
72 |
66 |
1.96 |
15.15 |
81.412 |
3.209 |
T P |
| 2ae6_A |
148 |
72 |
64 |
2.00 |
10.94 |
80.669 |
3.044 |
T P |
| 1z4e_A |
150 |
72 |
64 |
1.90 |
7.81 |
80.246 |
3.199 |
T P |
| 3ec4_A |
224 |
72 |
62 |
1.88 |
12.90 |
79.735 |
3.137 |
T P |
| 1i12_D |
157 |
72 |
61 |
1.66 |
9.84 |
79.697 |
3.471 |
T P |
| 1vkc_A |
149 |
72 |
62 |
1.68 |
16.13 |
79.617 |
3.475 |
T P |
| 3fb3_B |
149 |
72 |
62 |
1.73 |
11.29 |
79.502 |
3.382 |
T P |
| 1i1d_D |
161 |
72 |
61 |
1.85 |
6.56 |
78.418 |
3.136 |
T P |
| 2b5g_B |
159 |
72 |
60 |
1.67 |
11.67 |
78.054 |
3.388 |
T P |
| 1yx0_A |
151 |
72 |
58 |
1.59 |
12.07 |
77.558 |
3.437 |
T P |
| 2b3u_B |
164 |
72 |
59 |
1.56 |
11.86 |
77.144 |
3.552 |
T P |
| 2fiw_A |
160 |
72 |
60 |
1.59 |
11.67 |
76.961 |
3.550 |
T P |
| 3bj7_A |
162 |
72 |
60 |
1.71 |
11.67 |
76.791 |
3.323 |
T P |
| 2fe7_B |
166 |
72 |
60 |
1.76 |
15.00 |
76.552 |
3.227 |
T P |
| 2fxf_A |
164 |
72 |
59 |
1.60 |
10.17 |
75.256 |
3.470 |
T P |
| 2g3t_A |
169 |
72 |
58 |
1.82 |
12.07 |
74.018 |
3.027 |
T P |
| 1i21_B |
157 |
72 |
61 |
2.01 |
6.56 |
73.002 |
2.890 |
T P |
| 2b5g_A |
164 |
72 |
59 |
2.06 |
10.17 |
72.470 |
2.733 |
T P |
| 2jev_A |
169 |
72 |
58 |
1.92 |
12.07 |
72.260 |
2.878 |
T P |
| 1u6m_A |
189 |
72 |
61 |
2.20 |
14.75 |
71.648 |
2.656 |
T P |
| 1q2y_A |
140 |
72 |
63 |
2.12 |
9.52 |
70.962 |
2.837 |
T P |
| 2bei_B |
159 |
72 |
56 |
1.86 |
12.50 |
70.631 |
2.861 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]