LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_21.5wLII_10601_4
Total number of 3D structures: 49
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
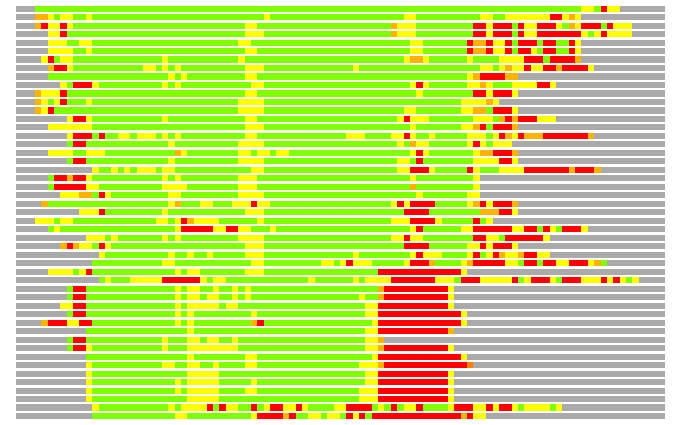
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3f6v_A |
96 |
102 |
91 |
0.68 |
24.18 |
88.390 |
11.667 |
T P |
| 3f6o_A |
97 |
102 |
84 |
1.84 |
16.67 |
75.486 |
4.334 |
T P |
| 2cwe_A |
191 |
102 |
79 |
1.75 |
17.72 |
72.467 |
4.262 |
T P |
| 1uly_A |
190 |
102 |
77 |
1.66 |
18.18 |
72.115 |
4.378 |
T P |
| 1xnp_B |
199 |
102 |
75 |
1.52 |
14.67 |
71.070 |
4.628 |
T P |
| 2p4w_B |
198 |
102 |
76 |
1.55 |
15.79 |
70.859 |
4.615 |
T P |
| 2oqg_A |
109 |
102 |
77 |
1.74 |
28.57 |
70.301 |
4.177 |
T P |
| 1ub9_A |
100 |
102 |
77 |
2.07 |
19.48 |
68.528 |
3.549 |
T P |
| 3cuo_A |
98 |
102 |
70 |
1.45 |
30.00 |
67.247 |
4.512 |
T P |
| 2d1h_A |
102 |
102 |
72 |
1.65 |
19.44 |
67.045 |
4.120 |
T P |
| 1u2w_B |
107 |
102 |
70 |
1.44 |
22.86 |
66.469 |
4.558 |
T P |
| 1r22_B |
97 |
102 |
72 |
1.71 |
30.56 |
66.434 |
3.980 |
T P |
| 1r1u_C |
98 |
102 |
72 |
1.74 |
34.72 |
66.366 |
3.908 |
T P |
| 3bdd_A |
140 |
102 |
70 |
1.81 |
12.86 |
64.143 |
3.673 |
T P |
| 1r1t_B |
99 |
102 |
70 |
1.60 |
31.43 |
63.751 |
4.110 |
T P |
| 2a61_A |
142 |
102 |
69 |
2.26 |
14.49 |
63.623 |
2.927 |
T P |
| 1sfx_A |
109 |
102 |
67 |
1.62 |
26.87 |
62.964 |
3.884 |
T P |
| 3df8_A |
109 |
102 |
70 |
1.95 |
17.14 |
62.717 |
3.415 |
T P |
| 3e6m_D |
149 |
102 |
66 |
1.52 |
18.18 |
61.846 |
4.063 |
T P |
| 1jmr_B |
247 |
102 |
66 |
1.83 |
25.76 |
60.237 |
3.425 |
T P |
| 2fe3_B |
143 |
102 |
64 |
1.45 |
18.75 |
60.217 |
4.120 |
T P |
| 2rgv_B |
140 |
102 |
63 |
1.53 |
19.05 |
59.249 |
3.861 |
T P |
| 1q1h_A |
85 |
102 |
65 |
1.78 |
18.46 |
59.082 |
3.451 |
T P |
| 2jsc_B |
97 |
102 |
65 |
1.96 |
32.31 |
58.901 |
3.158 |
T P |
| 2pex_B |
137 |
102 |
62 |
1.71 |
20.97 |
58.523 |
3.419 |
T P |
| 1y0u_A |
89 |
102 |
66 |
1.90 |
21.21 |
58.076 |
3.293 |
T P |
| 2qlz_B |
220 |
102 |
65 |
1.67 |
15.38 |
57.886 |
3.663 |
T P |
| 1ku9_A |
151 |
102 |
64 |
1.75 |
12.50 |
57.671 |
3.468 |
T P |
| 2pfb_A |
129 |
102 |
62 |
1.65 |
19.35 |
57.611 |
3.542 |
T P |
| 3cta_A |
187 |
102 |
66 |
2.04 |
13.64 |
56.733 |
3.081 |
T P |
| 1bia_A |
292 |
102 |
65 |
1.98 |
13.85 |
53.396 |
3.122 |
T P |
| 2p5v_G |
158 |
102 |
52 |
1.66 |
17.31 |
47.561 |
2.954 |
T P |
| 2hoe_A |
354 |
102 |
60 |
2.30 |
13.33 |
45.343 |
2.498 |
T P |
| 2vbw_A |
147 |
102 |
49 |
1.55 |
20.41 |
45.240 |
2.964 |
T P |
| 2ivm_A |
147 |
102 |
49 |
1.58 |
20.41 |
44.965 |
2.915 |
T P |
| 2cyy_A |
151 |
102 |
49 |
1.65 |
12.24 |
44.941 |
2.800 |
T P |
| 2ia0_A |
156 |
102 |
48 |
1.31 |
14.58 |
44.874 |
3.398 |
T P |
| 2cfx_A |
140 |
102 |
48 |
1.54 |
18.75 |
44.728 |
2.930 |
T P |
| 1i1g_A |
140 |
102 |
47 |
1.36 |
12.77 |
44.367 |
3.223 |
T P |
| 2e1c_A |
147 |
102 |
48 |
1.54 |
12.50 |
44.258 |
2.924 |
T P |
| 2cg4_A |
149 |
102 |
49 |
1.78 |
12.24 |
44.083 |
2.611 |
T P |
| 2qz8_A |
146 |
102 |
47 |
1.42 |
21.28 |
43.761 |
3.089 |
T P |
| 2yx4_A |
150 |
102 |
48 |
1.83 |
18.75 |
43.498 |
2.487 |
T P |
| 2efp_A |
150 |
102 |
47 |
1.53 |
17.02 |
43.352 |
2.879 |
T P |
| 2e7x_A |
150 |
102 |
47 |
1.55 |
17.02 |
43.275 |
2.841 |
T P |
| 2pn6_A |
150 |
102 |
47 |
1.55 |
19.15 |
43.271 |
2.852 |
T P |
| 2pmh_A |
150 |
102 |
47 |
1.60 |
17.02 |
42.709 |
2.771 |
T P |
| 1v4r_A |
100 |
102 |
56 |
2.34 |
14.29 |
38.586 |
2.295 |
T P |
| 6pax_A |
133 |
102 |
40 |
1.99 |
12.50 |
33.208 |
1.915 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]