LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_210.5wLII_11184_35
Total number of 3D structures: 56
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
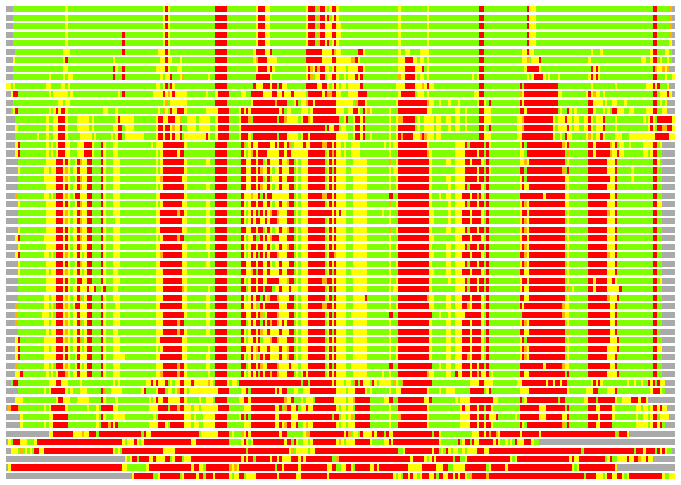
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1e1z_P |
481 |
281 |
256 |
1.02 |
25.00 |
89.205 |
22.861 |
T P |
| 1e3c_P |
481 |
281 |
256 |
1.02 |
25.39 |
89.202 |
22.776 |
T P |
| 1e2s_P |
481 |
281 |
256 |
1.04 |
24.61 |
88.998 |
22.542 |
T P |
| 1e33_P |
480 |
281 |
255 |
1.03 |
25.10 |
88.659 |
22.604 |
T P |
| 1auk_A |
480 |
281 |
255 |
1.04 |
24.71 |
88.498 |
22.363 |
T P |
| 1p49_A |
548 |
281 |
249 |
1.37 |
20.08 |
84.307 |
16.965 |
T P |
| 3ed4_B |
474 |
281 |
248 |
1.52 |
21.37 |
83.096 |
15.297 |
T P |
| 1fsu_A |
474 |
281 |
241 |
1.55 |
17.84 |
81.615 |
14.575 |
T P |
| 1hdh_A |
524 |
281 |
243 |
1.58 |
18.11 |
81.468 |
14.463 |
T P |
| 2qzu_A |
465 |
281 |
232 |
1.70 |
15.52 |
75.813 |
12.916 |
T P |
| 2vqr_A |
511 |
281 |
220 |
1.98 |
15.91 |
58.971 |
10.564 |
T P |
| 3b5q_B |
467 |
281 |
208 |
1.76 |
14.42 |
58.535 |
11.156 |
T P |
| 2gso_A |
382 |
281 |
199 |
1.91 |
13.07 |
52.936 |
9.892 |
T P |
| 2w5q_A |
424 |
281 |
202 |
2.17 |
13.86 |
48.944 |
8.879 |
T P |
| 2w5r_A |
424 |
281 |
191 |
2.11 |
14.14 |
47.263 |
8.628 |
T P |
| 2w8d_B |
421 |
281 |
199 |
2.28 |
14.07 |
47.153 |
8.374 |
T P |
| 2iuc_A |
340 |
281 |
182 |
1.95 |
12.64 |
47.153 |
8.867 |
T P |
| 2iuc_B |
342 |
281 |
185 |
1.95 |
12.43 |
46.927 |
9.025 |
T P |
| 1hjk_A |
449 |
281 |
184 |
2.02 |
15.22 |
46.140 |
8.680 |
T P |
| 1elx_A |
449 |
281 |
182 |
1.99 |
15.93 |
45.735 |
8.694 |
T P |
| 1ely_A |
449 |
281 |
181 |
1.98 |
15.47 |
45.722 |
8.712 |
T P |
| 1ali_A |
446 |
281 |
184 |
2.00 |
16.30 |
45.673 |
8.756 |
T P |
| 3bdf_A |
434 |
281 |
181 |
2.07 |
16.02 |
45.570 |
8.351 |
T P |
| 1ani_A |
446 |
281 |
185 |
2.03 |
15.68 |
45.559 |
8.686 |
T P |
| 1anh_A |
449 |
281 |
179 |
2.03 |
15.64 |
45.537 |
8.423 |
T P |
| 1ajb_A |
449 |
281 |
182 |
2.04 |
15.93 |
45.533 |
8.485 |
T P |
| 1anj_A |
446 |
281 |
183 |
1.98 |
15.85 |
45.518 |
8.781 |
T P |
| 1alk_A |
449 |
281 |
182 |
2.02 |
15.38 |
45.474 |
8.567 |
T P |
| 1hqa_A |
446 |
281 |
184 |
2.01 |
15.76 |
45.413 |
8.725 |
T P |
| 1khj_A |
444 |
281 |
182 |
2.07 |
15.38 |
45.380 |
8.382 |
T P |
| 1ura_A |
446 |
281 |
183 |
1.99 |
15.85 |
45.379 |
8.738 |
T P |
| 3dyc_A |
446 |
281 |
183 |
2.03 |
15.30 |
45.365 |
8.586 |
T P |
| 1y6v_A |
449 |
281 |
181 |
2.04 |
16.02 |
45.359 |
8.447 |
T P |
| 2anh_A |
446 |
281 |
184 |
2.08 |
15.22 |
45.326 |
8.451 |
T P |
| 1y7a_A |
449 |
281 |
183 |
2.10 |
15.30 |
45.276 |
8.330 |
T P |
| 1alh_A |
446 |
281 |
180 |
2.06 |
15.56 |
45.259 |
8.331 |
T P |
| 3bdh_A |
449 |
281 |
178 |
2.03 |
16.85 |
45.240 |
8.355 |
T P |
| 2g9y_A |
449 |
281 |
181 |
2.06 |
14.92 |
45.220 |
8.374 |
T P |
| 3cmr_A |
445 |
281 |
183 |
2.06 |
15.85 |
45.174 |
8.485 |
T P |
| 1kh7_A |
444 |
281 |
183 |
2.05 |
15.85 |
45.173 |
8.498 |
T P |
| 1kh5_A |
444 |
281 |
185 |
2.06 |
15.68 |
45.099 |
8.557 |
T P |
| 1b8j_A |
448 |
281 |
185 |
2.07 |
15.14 |
45.067 |
8.531 |
T P |
| 3bdg_B |
433 |
281 |
180 |
2.07 |
16.11 |
45.041 |
8.312 |
T P |
| 1elz_A |
449 |
281 |
181 |
2.11 |
14.92 |
44.649 |
8.189 |
T P |
| 2d1g_A |
481 |
281 |
184 |
2.17 |
16.85 |
44.468 |
8.091 |
T P |
| 2i09_B |
381 |
281 |
185 |
2.08 |
16.22 |
43.749 |
8.477 |
T P |
| 1ei6_A |
404 |
281 |
167 |
2.05 |
14.37 |
42.588 |
7.784 |
T P |
| 1o99_A |
509 |
281 |
170 |
2.02 |
18.24 |
42.212 |
8.001 |
T P |
| 2ify_A |
508 |
281 |
166 |
1.96 |
16.87 |
42.168 |
8.058 |
T P |
| 1o98_A |
509 |
281 |
169 |
1.99 |
18.93 |
42.113 |
8.080 |
T P |
| 1ti6_A |
875 |
281 |
81 |
2.65 |
7.41 |
19.537 |
2.950 |
T P |
| 1pjt_B |
455 |
281 |
81 |
2.46 |
9.88 |
18.045 |
3.163 |
T P |
| 1a4e_A |
488 |
281 |
62 |
2.59 |
9.68 |
14.028 |
2.309 |
T P |
| 2r85_A |
330 |
281 |
60 |
2.84 |
5.00 |
13.635 |
2.042 |
T P |
| 1s4n_A |
337 |
281 |
57 |
2.72 |
12.28 |
13.579 |
2.023 |
T P |
| 3fpx_A |
499 |
281 |
58 |
2.81 |
10.34 |
13.088 |
1.992 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]