LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_217.5wLII_11184_63
Total number of 3D structures: 70
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
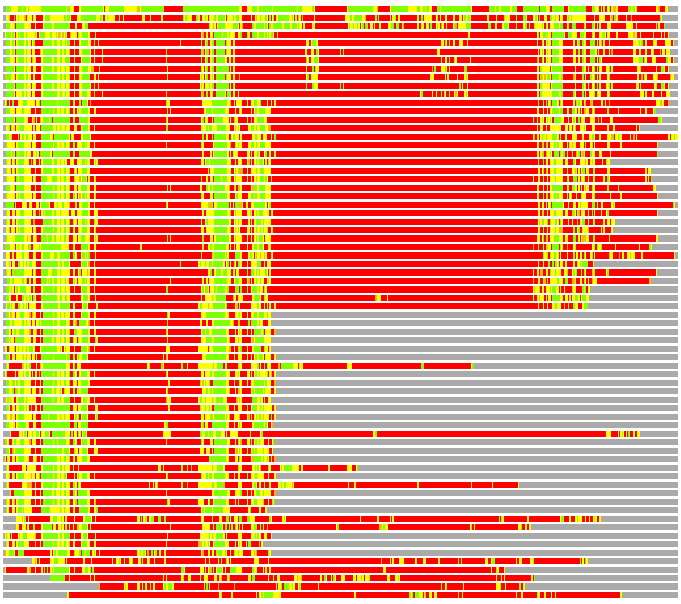
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2vsf_A |
588 |
514 |
386 |
1.74 |
17.88 |
68.949 |
20.932 |
T P |
| 3crv_A |
551 |
514 |
248 |
2.62 |
12.50 |
29.102 |
9.129 |
T P |
| 2vl7_A |
459 |
514 |
219 |
2.35 |
15.53 |
28.117 |
8.951 |
T P |
| 3bxz_B |
440 |
514 |
161 |
2.41 |
13.66 |
20.299 |
6.410 |
T P |
| 1d9x_A |
590 |
514 |
138 |
2.26 |
19.57 |
18.419 |
5.848 |
T P |
| 1t5l_A |
595 |
514 |
132 |
2.34 |
19.70 |
17.797 |
5.413 |
T P |
| 1d9z_A |
590 |
514 |
135 |
2.25 |
17.78 |
17.779 |
5.749 |
T P |
| 1d2m_A |
552 |
514 |
132 |
2.35 |
23.48 |
17.606 |
5.378 |
T P |
| 2fdc_B |
585 |
514 |
134 |
2.48 |
18.66 |
17.545 |
5.202 |
T P |
| 1c4o_A |
504 |
514 |
129 |
2.29 |
23.26 |
17.196 |
5.395 |
T P |
| 2d7d_A |
621 |
514 |
131 |
2.38 |
16.03 |
17.149 |
5.281 |
T P |
| 1wp9_A |
479 |
514 |
126 |
2.39 |
14.29 |
16.996 |
5.054 |
T P |
| 2j0s_A |
391 |
514 |
128 |
2.29 |
15.62 |
16.919 |
5.348 |
T P |
| 1z63_A |
468 |
514 |
122 |
2.25 |
16.39 |
16.686 |
5.189 |
T P |
| 2db3_A |
420 |
514 |
132 |
2.62 |
17.42 |
16.635 |
4.856 |
T P |
| 3dmq_A |
961 |
514 |
131 |
2.41 |
14.50 |
16.466 |
5.223 |
T P |
| 3g0h_A |
408 |
514 |
128 |
2.47 |
20.31 |
16.454 |
4.972 |
T P |
| 1z6a_A |
480 |
514 |
121 |
2.34 |
14.88 |
16.364 |
4.963 |
T P |
| 1hv8_A |
363 |
514 |
128 |
2.54 |
21.09 |
16.293 |
4.846 |
T P |
| 2vso_A |
366 |
514 |
125 |
2.49 |
18.40 |
16.279 |
4.822 |
T P |
| 1xtk_A |
382 |
514 |
126 |
2.43 |
18.25 |
16.262 |
4.978 |
T P |
| 2hyi_C |
392 |
514 |
129 |
2.36 |
15.50 |
16.221 |
5.237 |
T P |
| 3fht_A |
392 |
514 |
124 |
2.40 |
21.77 |
16.160 |
4.970 |
T P |
| 2v1x_A |
527 |
514 |
128 |
2.40 |
14.06 |
16.155 |
5.111 |
T P |
| 1xtj_A |
374 |
514 |
133 |
2.58 |
20.30 |
16.132 |
4.967 |
T P |
| 2z0m_A |
331 |
514 |
128 |
2.46 |
21.88 |
16.022 |
4.999 |
T P |
| 1s2m_A |
377 |
514 |
122 |
2.53 |
18.85 |
16.014 |
4.637 |
T P |
| 3eiq_D |
371 |
514 |
129 |
2.54 |
19.38 |
15.772 |
4.879 |
T P |
| 2gk6_A |
602 |
514 |
121 |
2.34 |
19.01 |
15.527 |
4.951 |
T P |
| 3ews_A |
416 |
514 |
120 |
2.48 |
17.50 |
15.484 |
4.645 |
T P |
| 2hjv_A |
158 |
514 |
112 |
2.26 |
9.82 |
15.318 |
4.740 |
T P |
| 1xti_A |
381 |
514 |
122 |
2.53 |
18.03 |
15.230 |
4.644 |
T P |
| 1fuu_B |
380 |
514 |
124 |
2.60 |
14.52 |
15.133 |
4.588 |
T P |
| 1oyw_A |
516 |
514 |
116 |
2.28 |
12.07 |
15.131 |
4.870 |
T P |
| 1gku_B |
1011 |
514 |
117 |
2.35 |
6.84 |
14.964 |
4.767 |
T P |
| 2zu6_C |
351 |
514 |
107 |
2.34 |
6.54 |
14.159 |
4.392 |
T P |
| 2oxc_A |
205 |
514 |
98 |
2.29 |
15.31 |
13.921 |
4.099 |
T P |
| 3b6e_A |
182 |
514 |
94 |
2.18 |
20.21 |
13.915 |
4.124 |
T P |
| 1qva_A |
213 |
514 |
99 |
2.32 |
21.21 |
13.887 |
4.098 |
T P |
| 2hxy_A |
376 |
514 |
99 |
2.35 |
15.15 |
13.712 |
4.044 |
T P |
| 2p6n_A |
160 |
514 |
95 |
2.23 |
9.47 |
13.588 |
4.077 |
T P |
| 2pl3_A |
232 |
514 |
94 |
2.21 |
18.09 |
13.409 |
4.078 |
T P |
| 2p6r_A |
683 |
514 |
105 |
2.55 |
13.33 |
13.269 |
3.966 |
T P |
| 1vec_A |
206 |
514 |
95 |
2.48 |
17.89 |
13.225 |
3.677 |
T P |
| 2j0u_B |
347 |
514 |
95 |
2.27 |
15.79 |
13.045 |
4.008 |
T P |
| 2j0u_A |
366 |
514 |
97 |
2.33 |
16.49 |
12.976 |
3.986 |
T P |
| 3eaq_B |
209 |
514 |
94 |
2.35 |
14.89 |
12.967 |
3.835 |
T P |
| 3ber_A |
220 |
514 |
96 |
2.37 |
19.79 |
12.901 |
3.894 |
T P |
| 1t6n_A |
207 |
514 |
98 |
2.39 |
18.37 |
12.816 |
3.939 |
T P |
| 3dkp_A |
240 |
514 |
89 |
2.07 |
20.22 |
12.705 |
4.104 |
T P |
| 1z3i_X |
644 |
514 |
97 |
2.53 |
12.37 |
12.606 |
3.690 |
T P |
| 3fhc_B |
224 |
514 |
89 |
2.31 |
24.72 |
12.597 |
3.697 |
T P |
| 1q0u_B |
210 |
514 |
92 |
2.27 |
20.65 |
12.558 |
3.888 |
T P |
| 1qde_A |
201 |
514 |
87 |
2.26 |
20.69 |
12.545 |
3.692 |
T P |
| 2va8_B |
695 |
514 |
98 |
2.50 |
12.24 |
12.528 |
3.773 |
T P |
| 3fe2_A |
234 |
514 |
93 |
2.32 |
19.35 |
12.503 |
3.841 |
T P |
| 2zj8_A |
700 |
514 |
93 |
2.62 |
11.83 |
12.121 |
3.421 |
T P |
| 3bor_A |
194 |
514 |
86 |
2.29 |
23.26 |
12.115 |
3.601 |
T P |
| 2g9n_A |
216 |
514 |
89 |
2.33 |
13.48 |
12.069 |
3.666 |
T P |
| 2gxq_A |
206 |
514 |
78 |
2.28 |
24.36 |
11.557 |
3.281 |
T P |
| 1gl9_B |
1020 |
514 |
91 |
2.69 |
12.09 |
11.521 |
3.267 |
T P |
| 1oyy_A |
512 |
514 |
85 |
2.59 |
8.24 |
11.470 |
3.165 |
T P |
| 2jgn_B |
161 |
514 |
84 |
2.29 |
10.71 |
11.204 |
3.508 |
T P |
| 1wrb_A |
232 |
514 |
79 |
2.38 |
18.99 |
10.954 |
3.186 |
T P |
| 2i4i_A |
408 |
514 |
85 |
2.48 |
7.06 |
10.731 |
3.290 |
T P |
| 1gm5_A |
729 |
514 |
77 |
2.84 |
5.19 |
9.712 |
2.615 |
T P |
| 2ffh_A |
407 |
514 |
72 |
2.57 |
9.72 |
9.563 |
2.695 |
T P |
| 1qvr_A |
803 |
514 |
75 |
2.83 |
6.67 |
9.538 |
2.558 |
T P |
| 2v7k_A |
345 |
514 |
52 |
2.70 |
7.69 |
7.096 |
1.858 |
T P |
| 2a6h_A |
229 |
514 |
50 |
2.78 |
6.00 |
6.498 |
1.738 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]