LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_221.5wLII_11185_11
Total number of 3D structures: 90
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
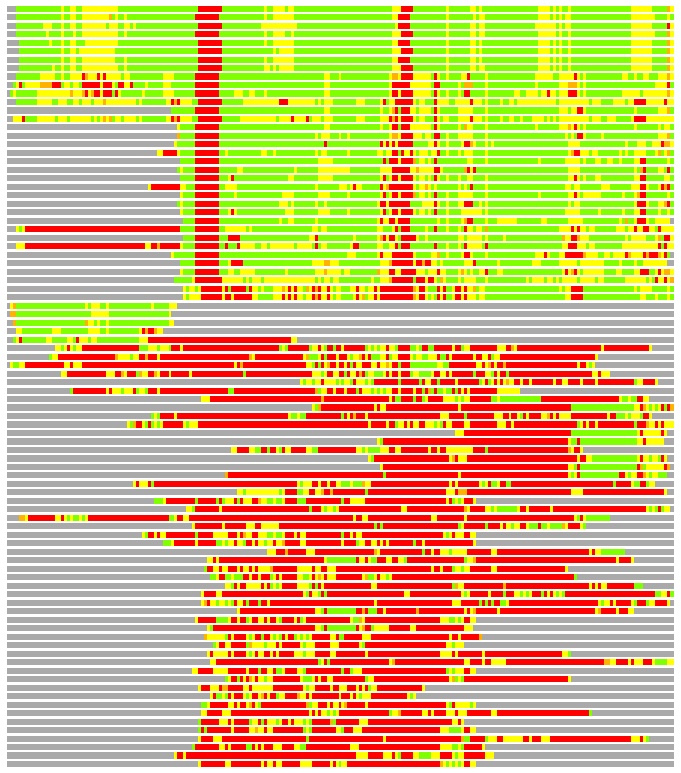
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1qpz_A |
339 |
223 |
208 |
1.65 |
17.31 |
86.838 |
11.889 |
T P |
| 1jft_A |
340 |
223 |
208 |
1.66 |
17.79 |
86.771 |
11.843 |
T P |
| 1jh9_A |
339 |
223 |
207 |
1.70 |
17.39 |
86.628 |
11.511 |
T P |
| 1jfs_A |
339 |
223 |
208 |
1.69 |
17.79 |
86.331 |
11.632 |
T P |
| 2puc_A |
338 |
223 |
207 |
1.66 |
17.39 |
86.309 |
11.767 |
T P |
| 1bdh_A |
338 |
223 |
207 |
1.65 |
17.39 |
86.130 |
11.859 |
T P |
| 1vpw_A |
338 |
223 |
207 |
1.67 |
17.39 |
86.014 |
11.682 |
T P |
| 2pue_A |
338 |
223 |
207 |
1.69 |
17.39 |
85.878 |
11.553 |
T P |
| 1zvv_A |
332 |
223 |
202 |
2.02 |
11.88 |
77.032 |
9.526 |
T P |
| 1rzr_G |
332 |
223 |
188 |
2.01 |
10.11 |
72.609 |
8.910 |
T P |
| 1rzr_D |
332 |
223 |
194 |
2.14 |
10.82 |
71.694 |
8.649 |
T P |
| 2pe5_B |
330 |
223 |
193 |
2.40 |
13.47 |
64.798 |
7.722 |
T P |
| 2nzu_G |
275 |
223 |
153 |
1.63 |
8.50 |
63.142 |
8.841 |
T P |
| 1efa_B |
330 |
223 |
196 |
2.34 |
12.76 |
63.133 |
8.046 |
T P |
| 2fep_A |
273 |
223 |
151 |
1.70 |
6.62 |
62.275 |
8.380 |
T P |
| 1sxh_A |
273 |
223 |
150 |
1.69 |
9.33 |
61.919 |
8.384 |
T P |
| 3egc_D |
262 |
223 |
146 |
1.57 |
12.33 |
61.187 |
8.721 |
T P |
| 2o20_B |
275 |
223 |
153 |
1.94 |
9.80 |
61.076 |
7.486 |
T P |
| 1jye_A |
271 |
223 |
146 |
1.67 |
10.96 |
59.785 |
8.232 |
T P |
| 2rgy_A |
275 |
223 |
146 |
1.79 |
7.53 |
59.695 |
7.713 |
T P |
| 3edc_A |
300 |
223 |
145 |
1.74 |
11.72 |
59.218 |
7.898 |
T P |
| 3c3k_B |
272 |
223 |
143 |
1.75 |
9.09 |
59.096 |
7.723 |
T P |
| 1jyf_A |
272 |
223 |
146 |
1.82 |
11.64 |
58.835 |
7.589 |
T P |
| 3ctp_A |
266 |
223 |
141 |
1.51 |
9.93 |
58.792 |
8.751 |
T P |
| 1lbi_A |
296 |
223 |
146 |
1.78 |
11.64 |
58.713 |
7.769 |
T P |
| 3bil_A |
257 |
223 |
143 |
1.75 |
11.89 |
58.308 |
7.737 |
T P |
| 2jcg_A |
322 |
223 |
148 |
1.95 |
8.78 |
57.826 |
7.218 |
T P |
| 3clk_B |
262 |
223 |
139 |
1.81 |
9.35 |
56.306 |
7.265 |
T P |
| 2hsg_A |
328 |
223 |
146 |
2.09 |
8.90 |
52.782 |
6.670 |
T P |
| 2iks_A |
276 |
223 |
136 |
1.95 |
8.09 |
51.269 |
6.627 |
T P |
| 3brq_B |
277 |
223 |
139 |
2.03 |
10.07 |
48.762 |
6.539 |
T P |
| 3e3m_A |
270 |
223 |
142 |
2.15 |
4.93 |
48.368 |
6.303 |
T P |
| 3dbi_A |
278 |
223 |
144 |
2.42 |
7.64 |
43.334 |
5.707 |
T P |
| 1jhz_A |
276 |
223 |
121 |
2.19 |
9.92 |
41.870 |
5.279 |
T P |
| 1dbq_A |
276 |
223 |
119 |
2.15 |
10.08 |
41.529 |
5.298 |
T P |
| 1l1m_A |
62 |
223 |
56 |
1.59 |
19.64 |
23.824 |
3.308 |
T P |
| 1cjg_A |
62 |
223 |
54 |
1.56 |
22.22 |
23.240 |
3.249 |
T P |
| 2bjc_A |
62 |
223 |
54 |
1.60 |
20.37 |
22.838 |
3.168 |
T P |
| 1lqc_A |
56 |
223 |
46 |
1.93 |
21.74 |
18.693 |
2.262 |
T P |
| 1pru_A |
56 |
223 |
45 |
2.02 |
15.56 |
17.912 |
2.125 |
T P |
| 2axz_A |
306 |
223 |
57 |
2.51 |
10.53 |
17.081 |
2.182 |
T P |
| 2axv_B |
303 |
223 |
56 |
2.52 |
14.29 |
16.995 |
2.136 |
T P |
| 2aw6_A |
287 |
223 |
53 |
2.71 |
9.43 |
16.494 |
1.884 |
T P |
| 2awi_A |
298 |
223 |
58 |
2.80 |
6.90 |
16.088 |
1.999 |
T P |
| 2axu_A |
303 |
223 |
49 |
2.55 |
6.12 |
15.561 |
1.848 |
T P |
| 2grm_A |
315 |
223 |
54 |
2.78 |
12.96 |
15.496 |
1.873 |
T P |
| 2ras_B |
204 |
223 |
44 |
2.23 |
11.36 |
15.157 |
1.886 |
T P |
| 2p5t_E |
95 |
223 |
38 |
2.20 |
7.89 |
14.410 |
1.649 |
T P |
| 2bnm_A |
194 |
223 |
44 |
2.72 |
4.55 |
13.531 |
1.563 |
T P |
| 2qfc_A |
284 |
223 |
48 |
3.01 |
10.42 |
13.487 |
1.543 |
T P |
| 2b5a_A |
77 |
223 |
33 |
1.94 |
3.03 |
13.188 |
1.620 |
T P |
| 1y7y_A |
69 |
223 |
32 |
2.00 |
3.12 |
12.889 |
1.523 |
T P |
| 3es1_A |
163 |
223 |
43 |
2.81 |
4.65 |
12.757 |
1.477 |
T P |
| 3f6w_A |
82 |
223 |
33 |
2.37 |
12.12 |
12.206 |
1.335 |
T P |
| 2ewt_A |
71 |
223 |
32 |
2.15 |
0.00 |
12.137 |
1.423 |
T P |
| 2ofy_A |
82 |
223 |
31 |
2.05 |
3.23 |
12.091 |
1.443 |
T P |
| 3bdn_A |
234 |
223 |
41 |
2.49 |
7.32 |
12.054 |
1.585 |
T P |
| 3eus_A |
85 |
223 |
36 |
2.98 |
5.56 |
11.362 |
1.168 |
T P |
| 1rio_A |
97 |
223 |
38 |
2.55 |
5.26 |
11.239 |
1.433 |
T P |
| 2iuo_A |
156 |
223 |
36 |
2.48 |
2.78 |
11.071 |
1.395 |
T P |
| 2eby_A |
102 |
223 |
35 |
2.48 |
5.71 |
10.976 |
1.357 |
T P |
| 1y9q_A |
178 |
223 |
37 |
2.63 |
10.81 |
10.880 |
1.354 |
T P |
| 1lli_B |
92 |
223 |
39 |
2.81 |
7.69 |
10.750 |
1.338 |
T P |
| 1lmb_4 |
92 |
223 |
36 |
2.75 |
2.78 |
10.735 |
1.263 |
T P |
| 3cec_A |
91 |
223 |
33 |
2.74 |
6.06 |
10.511 |
1.161 |
T P |
| 2ivb_A |
156 |
223 |
30 |
2.41 |
10.00 |
10.488 |
1.195 |
T P |
| 1x57_A |
91 |
223 |
35 |
2.91 |
5.71 |
10.424 |
1.165 |
T P |
| 2jvl_A |
107 |
223 |
35 |
2.55 |
14.29 |
10.361 |
1.323 |
T P |
| 1b0n_A |
103 |
223 |
35 |
2.76 |
8.57 |
10.029 |
1.224 |
T P |
| 2ivg_A |
156 |
223 |
33 |
2.46 |
6.06 |
9.997 |
1.290 |
T P |
| 2k9q_A |
77 |
223 |
36 |
2.67 |
2.78 |
9.966 |
1.301 |
T P |
| 2iu7_A |
156 |
223 |
29 |
2.58 |
6.90 |
9.937 |
1.081 |
T P |
| 3b7h_A |
76 |
223 |
32 |
2.61 |
15.62 |
9.831 |
1.181 |
T P |
| 2ivq_A |
156 |
223 |
28 |
2.56 |
7.14 |
9.764 |
1.054 |
T P |
| 3dnv_B |
71 |
223 |
34 |
2.87 |
11.76 |
9.634 |
1.145 |
T P |
| 1r63_A |
63 |
223 |
31 |
2.78 |
0.00 |
9.559 |
1.076 |
T P |
| 1adr_A |
76 |
223 |
33 |
2.92 |
0.00 |
9.372 |
1.092 |
T P |
| 1dw9_A |
156 |
223 |
30 |
2.81 |
3.33 |
9.243 |
1.030 |
T P |
| 3bs3_A |
62 |
223 |
30 |
2.73 |
3.33 |
9.083 |
1.060 |
T P |
| 2r63_A |
63 |
223 |
28 |
2.60 |
7.14 |
9.024 |
1.038 |
T P |
| 1r69_A |
63 |
223 |
29 |
3.03 |
10.34 |
8.953 |
0.928 |
T P |
| 2r1j_L |
66 |
223 |
29 |
2.78 |
13.79 |
8.885 |
1.005 |
T P |
| 1utx_A |
66 |
223 |
29 |
2.82 |
6.90 |
8.869 |
0.993 |
T P |
| 3clc_B |
77 |
223 |
29 |
2.88 |
3.45 |
8.825 |
0.972 |
T P |
| 2cro_A |
65 |
223 |
29 |
2.84 |
3.45 |
8.737 |
0.988 |
T P |
| 3f52_E |
78 |
223 |
29 |
2.73 |
13.79 |
8.575 |
1.025 |
T P |
| 2iv1_A |
156 |
223 |
29 |
2.93 |
0.00 |
8.540 |
0.956 |
T P |
| 2ppx_A |
62 |
223 |
27 |
2.85 |
3.70 |
8.539 |
0.914 |
T P |
| 2ef8_A |
84 |
223 |
27 |
2.68 |
14.81 |
8.467 |
0.972 |
T P |
| 1sq8_A |
64 |
223 |
27 |
2.86 |
7.41 |
8.018 |
0.913 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]