LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_228.5wLII_11195_20
Total number of 3D structures: 38
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
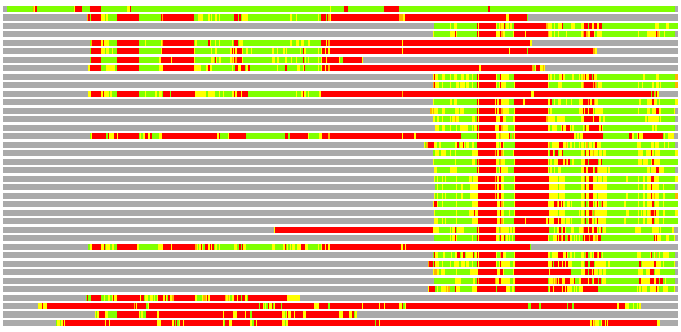
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3eo7_A |
487 |
522 |
486 |
0.47 |
39.30 |
92.608 |
85.565 |
T P |
| 3gbh_B |
213 |
522 |
136 |
1.83 |
16.18 |
23.734 |
7.041 |
T P |
| 3g14_B |
179 |
522 |
138 |
1.82 |
16.67 |
23.671 |
7.189 |
T P |
| 1bkj_A |
230 |
522 |
141 |
1.90 |
24.11 |
23.575 |
7.052 |
T P |
| 3ge6_A |
210 |
522 |
131 |
1.73 |
17.56 |
22.860 |
7.151 |
T P |
| 3eof_A |
248 |
522 |
131 |
1.84 |
19.85 |
22.452 |
6.739 |
T P |
| 1f5v_A |
240 |
522 |
128 |
1.72 |
15.62 |
22.412 |
7.025 |
T P |
| 3e10_B |
167 |
522 |
131 |
1.90 |
17.56 |
22.118 |
6.544 |
T P |
| 2b67_A |
201 |
522 |
142 |
1.85 |
17.61 |
21.714 |
7.282 |
T P |
| 3gag_A |
206 |
522 |
140 |
2.00 |
17.14 |
21.503 |
6.676 |
T P |
| 2fre_A |
200 |
522 |
127 |
1.84 |
16.54 |
21.446 |
6.532 |
T P |
| 1zch_A |
249 |
522 |
139 |
1.83 |
23.74 |
21.101 |
7.189 |
T P |
| 3bem_B |
210 |
522 |
141 |
2.06 |
17.73 |
20.980 |
6.541 |
T P |
| 3e39_A |
175 |
522 |
137 |
1.98 |
18.98 |
20.761 |
6.580 |
T P |
| 2r01_A |
195 |
522 |
132 |
1.86 |
14.39 |
20.541 |
6.738 |
T P |
| 1vkw_A |
217 |
522 |
148 |
2.06 |
10.81 |
20.353 |
6.858 |
T P |
| 1ywq_A |
199 |
522 |
143 |
2.11 |
11.19 |
20.024 |
6.462 |
T P |
| 2hay_D |
222 |
522 |
139 |
2.10 |
16.55 |
19.988 |
6.320 |
T P |
| 3ek3_A |
187 |
522 |
133 |
1.91 |
21.80 |
19.834 |
6.614 |
T P |
| 1nox_A |
200 |
522 |
137 |
2.00 |
21.90 |
19.708 |
6.516 |
T P |
| 1nec_A |
216 |
522 |
140 |
2.10 |
15.71 |
19.619 |
6.350 |
T P |
| 1yki_B |
217 |
522 |
139 |
2.09 |
17.27 |
19.577 |
6.335 |
T P |
| 1icv_A |
216 |
522 |
141 |
2.14 |
17.02 |
19.570 |
6.308 |
T P |
| 1vfr_A |
217 |
522 |
135 |
2.08 |
11.85 |
18.868 |
6.205 |
T P |
| 1kqb_A |
216 |
522 |
139 |
2.13 |
16.55 |
18.853 |
6.238 |
T P |
| 1v5y_A |
217 |
522 |
138 |
2.11 |
12.32 |
18.604 |
6.251 |
T P |
| 2isk_A |
219 |
522 |
137 |
2.16 |
18.25 |
18.427 |
6.062 |
T P |
| 3eo8_A |
219 |
522 |
131 |
2.03 |
16.03 |
18.290 |
6.143 |
T P |
| 1yw3_B |
201 |
522 |
127 |
2.07 |
13.39 |
18.121 |
5.863 |
T P |
| 2ifa_B |
201 |
522 |
132 |
2.17 |
12.12 |
17.972 |
5.816 |
T P |
| 2i7h_A |
187 |
522 |
131 |
2.28 |
14.50 |
17.774 |
5.508 |
T P |
| 2h0u_A |
196 |
522 |
120 |
2.05 |
14.17 |
17.340 |
5.576 |
T P |
| 3bm1_A |
177 |
522 |
120 |
2.15 |
17.50 |
16.777 |
5.336 |
T P |
| 3bm2_B |
166 |
522 |
116 |
2.17 |
18.10 |
16.324 |
5.116 |
T P |
| 1z5x_U |
193 |
522 |
64 |
2.63 |
9.38 |
8.220 |
2.341 |
T P |
| 1w6k_A |
725 |
522 |
51 |
2.61 |
11.76 |
6.484 |
1.882 |
T P |
| 1ft8_C |
244 |
522 |
44 |
2.65 |
9.09 |
5.658 |
1.602 |
T P |
| 1fo1_A |
215 |
522 |
43 |
2.82 |
13.95 |
5.365 |
1.474 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]