LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_235.5wLII_11195_46
Total number of 3D structures: 48
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
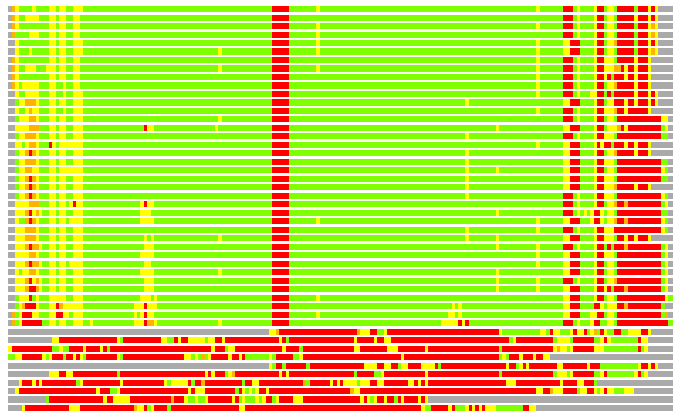
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3e7m_B |
421 |
196 |
172 |
1.17 |
16.28 |
85.171 |
13.546 |
T P |
| 4nos_A |
420 |
196 |
172 |
1.17 |
15.12 |
85.110 |
13.556 |
T P |
| 1m8d_A |
416 |
196 |
173 |
1.24 |
16.18 |
85.101 |
12.870 |
T P |
| 1df1_A |
420 |
196 |
173 |
1.26 |
16.18 |
84.976 |
12.716 |
T P |
| 1jwk_A |
413 |
196 |
171 |
1.17 |
15.79 |
84.741 |
13.413 |
T P |
| 1jwj_A |
412 |
196 |
172 |
1.25 |
15.70 |
84.638 |
12.736 |
T P |
| 1dwv_A |
420 |
196 |
171 |
1.14 |
16.37 |
84.392 |
13.771 |
T P |
| 2bhj_A |
414 |
196 |
173 |
1.36 |
16.18 |
84.325 |
11.815 |
T P |
| 1qom_A |
420 |
196 |
170 |
1.17 |
16.47 |
84.074 |
13.420 |
T P |
| 3dwj_B |
412 |
196 |
171 |
1.23 |
16.37 |
84.058 |
12.831 |
T P |
| 3e7g_C |
423 |
196 |
169 |
1.22 |
15.98 |
84.054 |
12.827 |
T P |
| 2g6m_B |
411 |
196 |
171 |
1.43 |
16.96 |
83.747 |
11.161 |
T P |
| 1n2n_A |
419 |
196 |
170 |
1.27 |
16.47 |
83.720 |
12.409 |
T P |
| 1nsi_A |
420 |
196 |
170 |
1.25 |
15.88 |
83.698 |
12.568 |
T P |
| 1zzt_A |
405 |
196 |
171 |
1.47 |
17.54 |
83.327 |
10.888 |
T P |
| 2g6j_B |
411 |
196 |
170 |
1.36 |
17.65 |
83.309 |
11.624 |
T P |
| 3ej8_A |
421 |
196 |
168 |
1.36 |
16.07 |
83.230 |
11.525 |
T P |
| 1lzx_B |
410 |
196 |
169 |
1.39 |
17.16 |
83.208 |
11.329 |
T P |
| 1zzq_B |
410 |
196 |
170 |
1.39 |
17.06 |
83.127 |
11.410 |
T P |
| 1vag_A |
420 |
196 |
170 |
1.35 |
17.06 |
83.096 |
11.722 |
T P |
| 1om4_B |
411 |
196 |
169 |
1.33 |
17.16 |
83.067 |
11.833 |
T P |
| 1p6i_B |
410 |
196 |
169 |
1.38 |
17.16 |
82.966 |
11.442 |
T P |
| 1zzu_B |
411 |
196 |
169 |
1.32 |
17.75 |
82.926 |
11.886 |
T P |
| 1zzs_A |
405 |
196 |
169 |
1.45 |
18.34 |
82.821 |
10.874 |
T P |
| 1q2o_A |
405 |
196 |
169 |
1.30 |
17.75 |
82.720 |
12.056 |
T P |
| 3e7s_A |
403 |
196 |
170 |
1.42 |
17.65 |
82.678 |
11.158 |
T P |
| 1p6k_B |
410 |
196 |
169 |
1.39 |
17.75 |
82.622 |
11.367 |
T P |
| 3nos_A |
400 |
196 |
171 |
1.47 |
18.71 |
82.590 |
10.870 |
T P |
| 1d0c_A |
416 |
196 |
168 |
1.38 |
18.45 |
82.514 |
11.384 |
T P |
| 3e7s_B |
409 |
196 |
170 |
1.40 |
17.65 |
82.438 |
11.360 |
T P |
| 1p6m_A |
404 |
196 |
169 |
1.38 |
17.75 |
82.338 |
11.402 |
T P |
| 1m9m_A |
400 |
196 |
170 |
1.43 |
18.82 |
82.308 |
11.116 |
T P |
| 2g6o_A |
416 |
196 |
169 |
1.40 |
17.75 |
82.282 |
11.285 |
T P |
| 1i83_A |
416 |
196 |
167 |
1.37 |
17.37 |
82.156 |
11.385 |
T P |
| 2flq_A |
359 |
196 |
169 |
1.37 |
17.75 |
81.921 |
11.530 |
T P |
| 2an2_A |
360 |
196 |
165 |
1.43 |
15.76 |
80.635 |
10.800 |
T P |
| 1m7v_A |
361 |
196 |
165 |
1.41 |
16.97 |
80.089 |
10.927 |
T P |
| 1mjt_B |
347 |
196 |
161 |
1.52 |
16.15 |
77.399 |
9.928 |
T P |
| 1vpx_E |
216 |
196 |
47 |
2.32 |
10.64 |
17.850 |
1.942 |
T P |
| 2hfw_A |
257 |
196 |
46 |
2.56 |
6.52 |
15.909 |
1.731 |
T P |
| 1z97_A |
263 |
196 |
43 |
2.48 |
0.00 |
15.779 |
1.668 |
T P |
| 2hfy_A |
259 |
196 |
43 |
2.46 |
2.33 |
14.776 |
1.681 |
T P |
| 2gjg_A |
247 |
196 |
39 |
2.48 |
12.82 |
14.314 |
1.511 |
T P |
| 2hfx_A |
259 |
196 |
39 |
2.46 |
0.00 |
14.261 |
1.523 |
T P |
| 1z93_A |
263 |
196 |
43 |
2.89 |
9.30 |
14.140 |
1.440 |
T P |
| 2rde_B |
225 |
196 |
39 |
2.78 |
2.56 |
13.733 |
1.355 |
T P |
| 1ywu_A |
125 |
196 |
38 |
2.58 |
2.63 |
12.912 |
1.415 |
T P |
| 1yln_A |
225 |
196 |
35 |
2.51 |
2.86 |
12.438 |
1.342 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]