LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_236.5wLII_11195_47
Total number of 3D structures: 109
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
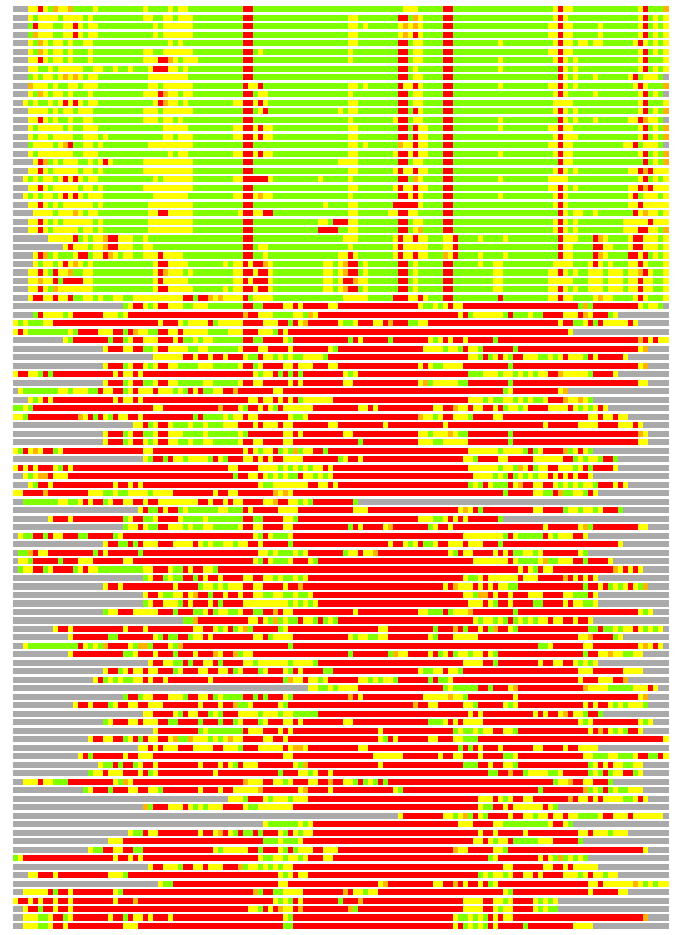
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2z1q_B |
549 |
131 |
121 |
1.48 |
18.18 |
87.003 |
7.636 |
T P |
| 1ivh_A |
387 |
131 |
120 |
1.60 |
17.50 |
84.090 |
7.065 |
T P |
| 3b96_A |
554 |
131 |
120 |
1.74 |
13.33 |
84.058 |
6.517 |
T P |
| 2uxw_A |
567 |
131 |
120 |
1.75 |
13.33 |
84.035 |
6.495 |
T P |
| 2d29_A |
386 |
131 |
120 |
1.61 |
13.33 |
83.933 |
6.998 |
T P |
| 2vig_E |
380 |
131 |
120 |
1.59 |
10.83 |
83.872 |
7.087 |
T P |
| 2dvl_A |
370 |
131 |
117 |
1.59 |
14.53 |
83.651 |
6.921 |
T P |
| 1rx0_A |
384 |
131 |
117 |
1.56 |
12.82 |
83.514 |
7.056 |
T P |
| 2ix6_A |
416 |
131 |
119 |
1.68 |
16.81 |
83.404 |
6.703 |
T P |
| 1buc_A |
383 |
131 |
120 |
1.72 |
12.50 |
83.270 |
6.579 |
T P |
| 1jqi_A |
384 |
131 |
118 |
1.68 |
10.17 |
83.055 |
6.645 |
T P |
| 2a1t_C |
388 |
131 |
118 |
1.79 |
14.41 |
83.052 |
6.248 |
T P |
| 2jif_A |
381 |
131 |
118 |
1.63 |
12.71 |
82.757 |
6.826 |
T P |
| 2ix5_A |
415 |
131 |
118 |
1.68 |
13.56 |
82.553 |
6.641 |
T P |
| 1udy_A |
385 |
131 |
118 |
1.71 |
11.86 |
82.500 |
6.524 |
T P |
| 3mdd_A |
385 |
131 |
118 |
1.74 |
11.86 |
82.466 |
6.408 |
T P |
| 2r0n_A |
390 |
131 |
119 |
1.84 |
14.29 |
82.396 |
6.126 |
T P |
| 1egd_A |
387 |
131 |
118 |
1.75 |
13.56 |
82.202 |
6.387 |
T P |
| 2pg0_A |
380 |
131 |
117 |
1.74 |
16.24 |
81.894 |
6.370 |
T P |
| 2r0m_A |
390 |
131 |
118 |
1.82 |
11.02 |
81.852 |
6.130 |
T P |
| 1ege_A |
387 |
131 |
117 |
1.75 |
13.68 |
81.679 |
6.324 |
T P |
| 1siq_A |
390 |
131 |
118 |
1.84 |
11.02 |
81.438 |
6.081 |
T P |
| 1ukw_A |
379 |
131 |
118 |
1.86 |
13.56 |
81.087 |
6.023 |
T P |
| 2eba_A |
380 |
131 |
116 |
1.81 |
13.79 |
80.046 |
6.076 |
T P |
| 1r2j_A |
353 |
131 |
115 |
1.83 |
14.78 |
79.731 |
5.946 |
T P |
| 3d6b_C |
377 |
131 |
116 |
1.83 |
12.93 |
79.647 |
6.014 |
T P |
| 3eom_A |
387 |
131 |
114 |
1.82 |
13.16 |
78.743 |
5.939 |
T P |
| 2fon_B |
656 |
131 |
112 |
1.88 |
10.71 |
75.153 |
5.660 |
T P |
| 1w07_B |
658 |
131 |
109 |
1.82 |
10.09 |
74.482 |
5.691 |
T P |
| 2ddh_A |
622 |
131 |
111 |
2.00 |
10.81 |
68.513 |
5.283 |
T P |
| 2c0u_A |
430 |
131 |
113 |
2.11 |
14.16 |
66.134 |
5.114 |
T P |
| 2reh_A |
430 |
131 |
113 |
2.02 |
13.27 |
65.768 |
5.326 |
T P |
| 2zaf_A |
430 |
131 |
111 |
2.03 |
12.61 |
65.517 |
5.203 |
T P |
| 2c12_A |
430 |
131 |
113 |
2.05 |
13.27 |
65.262 |
5.257 |
T P |
| 3djl_A |
538 |
131 |
110 |
2.24 |
10.91 |
62.556 |
4.702 |
T P |
| 1ny5_B |
385 |
131 |
43 |
2.30 |
13.95 |
23.545 |
1.791 |
T P |
| 3f9i_B |
229 |
131 |
47 |
2.68 |
8.51 |
23.004 |
1.690 |
T P |
| 1w25_A |
454 |
131 |
49 |
2.74 |
14.29 |
22.860 |
1.723 |
T P |
| 2nm0_A |
221 |
131 |
41 |
2.40 |
7.32 |
22.091 |
1.639 |
T P |
| 2ayz_A |
133 |
131 |
38 |
2.36 |
5.26 |
21.999 |
1.547 |
T P |
| 2chy_A |
128 |
131 |
40 |
2.58 |
17.50 |
21.912 |
1.491 |
T P |
| 3enn_B |
242 |
131 |
46 |
2.72 |
4.35 |
21.887 |
1.633 |
T P |
| 1mih_A |
128 |
131 |
41 |
2.58 |
12.20 |
21.832 |
1.529 |
T P |
| 1uzl_A |
231 |
131 |
43 |
2.62 |
0.00 |
21.609 |
1.579 |
T P |
| 1d4z_A |
128 |
131 |
40 |
2.46 |
17.50 |
21.512 |
1.565 |
T P |
| 1uzm_A |
227 |
131 |
41 |
2.50 |
4.88 |
21.493 |
1.578 |
T P |
| 2pd6_D |
242 |
131 |
43 |
2.72 |
2.33 |
21.438 |
1.524 |
T P |
| 2a8m_B |
313 |
131 |
44 |
2.84 |
2.27 |
21.300 |
1.496 |
T P |
| 2ayx_A |
254 |
131 |
43 |
2.69 |
0.00 |
21.113 |
1.541 |
T P |
| 1ymu_A |
125 |
131 |
37 |
2.80 |
10.81 |
20.959 |
1.275 |
T P |
| 2che_A |
128 |
131 |
40 |
2.51 |
15.00 |
20.899 |
1.531 |
T P |
| 1jbe_A |
126 |
131 |
39 |
2.49 |
12.82 |
20.805 |
1.505 |
T P |
| 1q7b_A |
243 |
131 |
41 |
2.72 |
7.32 |
20.717 |
1.456 |
T P |
| 1uls_B |
245 |
131 |
42 |
2.86 |
4.76 |
20.708 |
1.418 |
T P |
| 1rwb_A |
261 |
131 |
44 |
2.84 |
11.36 |
20.558 |
1.495 |
T P |
| 1ipe_A |
259 |
131 |
42 |
2.72 |
9.52 |
20.558 |
1.488 |
T P |
| 1fmc_A |
255 |
131 |
41 |
2.73 |
17.07 |
20.546 |
1.451 |
T P |
| 3eqv_A |
443 |
131 |
39 |
2.86 |
7.69 |
20.292 |
1.319 |
T P |
| 2a8j_B |
313 |
131 |
39 |
2.61 |
10.26 |
20.136 |
1.439 |
T P |
| 2hq1_A |
221 |
131 |
40 |
2.67 |
7.50 |
19.993 |
1.444 |
T P |
| 2r25_B |
133 |
131 |
34 |
2.24 |
8.82 |
19.821 |
1.452 |
T P |
| 1kmi_Y |
128 |
131 |
38 |
2.66 |
18.42 |
19.815 |
1.374 |
T P |
| 3emk_A |
241 |
131 |
38 |
2.33 |
7.89 |
19.693 |
1.563 |
T P |
| 2ntn_A |
218 |
131 |
38 |
2.71 |
7.89 |
19.664 |
1.351 |
T P |
| 2a4k_A |
237 |
131 |
38 |
2.69 |
13.16 |
19.621 |
1.362 |
T P |
| 2uvd_A |
246 |
131 |
39 |
2.56 |
7.69 |
19.604 |
1.469 |
T P |
| 2gwr_A |
225 |
131 |
36 |
2.43 |
0.00 |
19.385 |
1.423 |
T P |
| 1ab6_A |
125 |
131 |
37 |
2.65 |
2.70 |
19.368 |
1.347 |
T P |
| 2c07_A |
246 |
131 |
36 |
2.65 |
8.33 |
19.357 |
1.308 |
T P |
| 1ab5_A |
125 |
131 |
39 |
2.65 |
2.56 |
19.311 |
1.419 |
T P |
| 1udr_A |
126 |
131 |
38 |
2.57 |
13.16 |
19.287 |
1.422 |
T P |
| 1mvo_A |
121 |
131 |
38 |
2.63 |
10.53 |
19.161 |
1.394 |
T P |
| 2ae2_A |
259 |
131 |
36 |
2.55 |
5.56 |
19.120 |
1.361 |
T P |
| 1q7c_A |
243 |
131 |
38 |
2.57 |
10.53 |
19.106 |
1.423 |
T P |
| 1kgs_A |
219 |
131 |
38 |
2.84 |
2.63 |
19.074 |
1.293 |
T P |
| 2dtx_A |
255 |
131 |
34 |
2.59 |
5.88 |
19.061 |
1.266 |
T P |
| 1o5i_A |
234 |
131 |
39 |
2.76 |
12.82 |
19.001 |
1.363 |
T P |
| 2j8e_A |
128 |
131 |
37 |
2.73 |
8.11 |
18.981 |
1.305 |
T P |
| 2fka_A |
128 |
131 |
37 |
2.75 |
8.11 |
18.941 |
1.297 |
T P |
| 2a8l_B |
310 |
131 |
37 |
2.72 |
5.41 |
18.607 |
1.312 |
T P |
| 2cf2_E |
226 |
131 |
34 |
2.43 |
2.94 |
18.528 |
1.342 |
T P |
| 2a9r_A |
117 |
131 |
34 |
2.74 |
5.88 |
18.481 |
1.196 |
T P |
| 1vl8_B |
252 |
131 |
36 |
2.79 |
8.33 |
18.284 |
1.246 |
T P |
| 2a9o_A |
117 |
131 |
35 |
2.62 |
0.00 |
18.186 |
1.288 |
T P |
| 1ymv_A |
124 |
131 |
35 |
2.52 |
14.29 |
18.181 |
1.334 |
T P |
| 2pnf_A |
248 |
131 |
34 |
2.56 |
8.82 |
18.172 |
1.280 |
T P |
| 2a8i_A |
341 |
131 |
37 |
2.76 |
8.11 |
18.076 |
1.294 |
T P |
| 1tmy_A |
118 |
131 |
36 |
3.05 |
5.56 |
18.067 |
1.142 |
T P |
| 2ph3_A |
245 |
131 |
35 |
2.96 |
2.86 |
18.036 |
1.145 |
T P |
| 1u0s_Y |
118 |
131 |
34 |
2.40 |
2.94 |
17.371 |
1.361 |
T P |
| 2zk7_B |
246 |
131 |
35 |
2.69 |
0.00 |
17.248 |
1.255 |
T P |
| 1edo_A |
244 |
131 |
37 |
2.87 |
10.81 |
17.061 |
1.244 |
T P |
| 1geg_E |
256 |
131 |
33 |
2.85 |
3.03 |
17.055 |
1.120 |
T P |
| 1ys7_B |
226 |
131 |
33 |
2.80 |
3.03 |
16.737 |
1.139 |
T P |
| 1gee_A |
261 |
131 |
34 |
2.79 |
5.88 |
16.666 |
1.177 |
T P |
| 1ev7_A |
295 |
131 |
33 |
2.70 |
3.03 |
16.552 |
1.177 |
T P |
| 1mb3_A |
117 |
131 |
27 |
2.12 |
11.11 |
16.551 |
1.215 |
T P |
| 1oxk_B |
128 |
131 |
31 |
2.73 |
9.68 |
16.290 |
1.095 |
T P |
| 2iyn_B |
124 |
131 |
26 |
2.06 |
7.69 |
15.659 |
1.201 |
T P |
| 1dcf_A |
133 |
131 |
31 |
2.75 |
0.00 |
15.576 |
1.086 |
T P |
| 3c97_A |
120 |
131 |
28 |
2.61 |
10.71 |
15.474 |
1.033 |
T P |
| 2eub_A |
121 |
131 |
31 |
2.83 |
0.00 |
15.180 |
1.059 |
T P |
| 2ekp_A |
238 |
131 |
32 |
2.98 |
15.62 |
15.128 |
1.038 |
T P |
| 1ae1_B |
258 |
131 |
29 |
2.74 |
10.34 |
14.199 |
1.020 |
T P |
| 3ftp_A |
248 |
131 |
28 |
2.74 |
7.14 |
13.942 |
0.984 |
T P |
| 2z1n_B |
243 |
131 |
27 |
2.77 |
3.70 |
13.798 |
0.942 |
T P |
| 2d1y_C |
248 |
131 |
27 |
2.82 |
3.70 |
13.703 |
0.924 |
T P |
| 3chy_A |
128 |
131 |
27 |
2.72 |
7.41 |
13.309 |
0.957 |
T P |
| 1cye_A |
129 |
131 |
24 |
2.58 |
8.33 |
12.819 |
0.896 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]