LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_239.5wLII_11212_49
Total number of 3D structures: 37
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
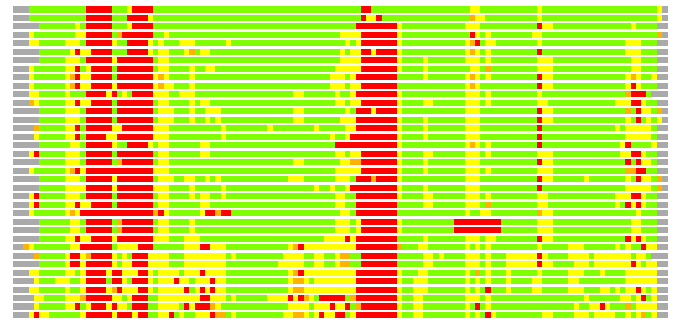
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2oby_A |
334 |
126 |
111 |
0.88 |
16.22 |
87.072 |
11.321 |
T P |
| 2j8z_A |
330 |
126 |
111 |
1.09 |
17.12 |
85.989 |
9.365 |
T P |
| 2eih_A |
343 |
126 |
101 |
1.22 |
10.89 |
78.227 |
7.671 |
T P |
| 3gaz_A |
331 |
126 |
103 |
1.51 |
10.68 |
76.995 |
6.396 |
T P |
| 1yb5_A |
324 |
126 |
104 |
1.69 |
8.65 |
75.961 |
5.804 |
T P |
| 1qor_A |
326 |
126 |
103 |
1.70 |
8.74 |
75.505 |
5.730 |
T P |
| 1rjw_A |
339 |
126 |
100 |
1.54 |
9.00 |
74.850 |
6.089 |
T P |
| 1llu_A |
341 |
126 |
102 |
1.73 |
5.88 |
73.956 |
5.585 |
T P |
| 2dfv_A |
346 |
126 |
100 |
1.77 |
9.00 |
73.879 |
5.359 |
T P |
| 2d8a_A |
333 |
126 |
100 |
1.77 |
9.00 |
73.775 |
5.337 |
T P |
| 2c0c_A |
353 |
126 |
100 |
1.71 |
13.00 |
73.282 |
5.524 |
T P |
| 1htb_A |
374 |
126 |
100 |
1.81 |
11.00 |
72.576 |
5.239 |
T P |
| 1nto_A |
347 |
126 |
99 |
1.74 |
8.08 |
72.370 |
5.382 |
T P |
| 2eer_B |
347 |
126 |
98 |
1.66 |
7.14 |
72.219 |
5.571 |
T P |
| 1pl8_A |
356 |
126 |
99 |
1.72 |
11.11 |
72.160 |
5.451 |
T P |
| 1pl6_A |
356 |
126 |
98 |
1.71 |
10.20 |
72.078 |
5.428 |
T P |
| 1wly_A |
322 |
126 |
96 |
1.53 |
12.50 |
72.052 |
5.902 |
T P |
| 1u3u_A |
374 |
126 |
99 |
1.78 |
11.11 |
72.030 |
5.259 |
T P |
| 1r37_A |
347 |
126 |
98 |
1.73 |
8.16 |
72.003 |
5.349 |
T P |
| 2hcy_A |
347 |
126 |
98 |
1.82 |
6.12 |
71.836 |
5.103 |
T P |
| 1jvb_A |
347 |
126 |
99 |
1.78 |
8.08 |
71.780 |
5.274 |
T P |
| 1e3j_A |
348 |
126 |
98 |
1.74 |
9.18 |
71.780 |
5.321 |
T P |
| 1u3w_A |
374 |
126 |
98 |
1.75 |
10.20 |
71.256 |
5.304 |
T P |
| 1ee2_A |
373 |
126 |
98 |
1.80 |
12.24 |
71.082 |
5.146 |
T P |
| 1xa0_A |
318 |
126 |
94 |
1.64 |
13.83 |
70.727 |
5.389 |
T P |
| 2cf2_D |
295 |
126 |
92 |
1.58 |
9.78 |
68.198 |
5.476 |
T P |
| 1iyz_A |
299 |
126 |
92 |
1.58 |
9.78 |
68.111 |
5.464 |
T P |
| 2dq4_A |
343 |
126 |
95 |
1.81 |
11.58 |
66.319 |
4.977 |
T P |
| 1vj1_A |
341 |
126 |
101 |
2.06 |
8.91 |
63.135 |
4.665 |
T P |
| 2vcy_A |
332 |
126 |
102 |
2.26 |
9.80 |
59.953 |
4.330 |
T P |
| 1zsy_A |
347 |
126 |
100 |
2.23 |
10.00 |
59.046 |
4.285 |
T P |
| 2w4q_A |
349 |
126 |
103 |
2.29 |
8.74 |
57.800 |
4.315 |
T P |
| 2zb3_A |
345 |
126 |
101 |
2.21 |
8.91 |
56.940 |
4.371 |
T P |
| 2zb4_A |
351 |
126 |
101 |
2.35 |
8.91 |
56.620 |
4.119 |
T P |
| 1zsv_A |
328 |
126 |
94 |
2.25 |
8.51 |
56.458 |
4.004 |
T P |
| 2j3h_B |
345 |
126 |
97 |
2.11 |
8.25 |
56.435 |
4.397 |
T P |
| 1v3v_A |
333 |
126 |
96 |
2.41 |
7.29 |
53.299 |
3.827 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]