LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_252.5wLII_11212_88
Total number of 3D structures: 36
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
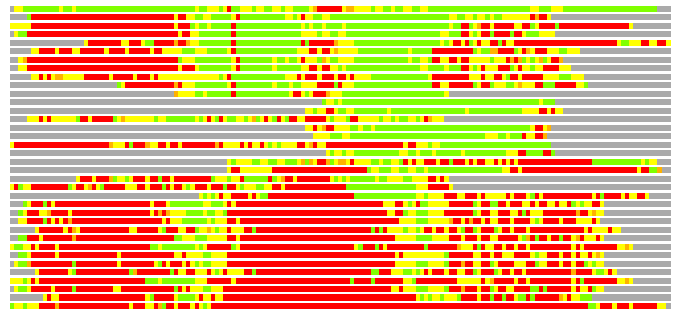
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1qvr_A |
803 |
161 |
150 |
1.72 |
16.00 |
85.703 |
8.245 |
T P |
| 1y1u_A |
544 |
161 |
86 |
1.95 |
8.14 |
47.813 |
4.195 |
T P |
| 2dq3_A |
425 |
161 |
81 |
1.87 |
7.41 |
45.693 |
4.114 |
T P |
| 2iw5_A |
666 |
161 |
82 |
1.83 |
10.98 |
44.940 |
4.250 |
T P |
| 1i0a_A |
453 |
161 |
81 |
2.02 |
4.94 |
43.503 |
3.814 |
T P |
| 2h94_A |
647 |
161 |
87 |
2.27 |
9.20 |
43.430 |
3.677 |
T P |
| 2v1d_A |
666 |
161 |
84 |
2.28 |
8.33 |
42.127 |
3.527 |
T P |
| 2hko_A |
647 |
161 |
79 |
2.02 |
11.39 |
41.320 |
3.724 |
T P |
| 2z3y_A |
643 |
161 |
88 |
2.58 |
9.09 |
40.768 |
3.279 |
T P |
| 2dw4_A |
634 |
161 |
79 |
2.07 |
11.39 |
40.368 |
3.642 |
T P |
| 1gax_A |
862 |
161 |
61 |
1.86 |
14.75 |
35.529 |
3.108 |
T P |
| 2e7s_D |
124 |
161 |
57 |
1.34 |
1.75 |
34.292 |
3.965 |
T P |
| 2ocy_A |
149 |
161 |
58 |
1.83 |
8.62 |
32.913 |
3.009 |
T P |
| 2inr_A |
442 |
161 |
79 |
2.65 |
12.66 |
32.091 |
2.869 |
T P |
| 1ei3_C |
397 |
161 |
55 |
1.87 |
7.27 |
31.244 |
2.789 |
T P |
| 2eqb_B |
93 |
161 |
54 |
1.76 |
7.41 |
30.999 |
2.898 |
T P |
| 1f5n_A |
570 |
161 |
65 |
2.44 |
7.69 |
30.362 |
2.564 |
T P |
| 1m1j_C |
390 |
161 |
52 |
1.77 |
7.69 |
29.928 |
2.784 |
T P |
| 1ng9_A |
794 |
161 |
68 |
2.53 |
11.76 |
28.433 |
2.589 |
T P |
| 2vzm_A |
395 |
161 |
49 |
2.08 |
6.12 |
25.002 |
2.245 |
T P |
| 1iok_A |
482 |
161 |
54 |
2.31 |
11.11 |
24.698 |
2.238 |
T P |
| 3cau_A |
526 |
161 |
52 |
2.38 |
9.62 |
24.068 |
2.100 |
T P |
| 1nj1_A |
463 |
161 |
51 |
2.21 |
3.92 |
23.930 |
2.204 |
T P |
| 1grl_A |
518 |
161 |
52 |
2.45 |
11.54 |
22.265 |
2.041 |
T P |
| 1kp8_A |
525 |
161 |
50 |
2.76 |
18.00 |
22.063 |
1.749 |
T P |
| 1oel_A |
524 |
161 |
50 |
2.76 |
18.00 |
21.360 |
1.750 |
T P |
| 1xck_A |
524 |
161 |
50 |
2.78 |
2.00 |
19.957 |
1.733 |
T P |
| 1ss8_A |
524 |
161 |
47 |
2.68 |
12.77 |
19.839 |
1.688 |
T P |
| 1pcq_A |
524 |
161 |
44 |
2.50 |
4.55 |
19.299 |
1.694 |
T P |
| 1j4z_A |
525 |
161 |
46 |
2.75 |
10.87 |
19.128 |
1.612 |
T P |
| 1sx3_A |
525 |
161 |
50 |
2.69 |
10.00 |
18.926 |
1.791 |
T P |
| 2eu1_A |
524 |
161 |
45 |
2.59 |
2.22 |
18.890 |
1.675 |
T P |
| 1we3_F |
529 |
161 |
43 |
2.52 |
13.95 |
18.733 |
1.641 |
T P |
| 1der_A |
525 |
161 |
46 |
2.67 |
8.70 |
18.538 |
1.661 |
T P |
| 3c9v_A |
526 |
161 |
40 |
2.56 |
7.50 |
17.048 |
1.503 |
T P |
| 2cd8_B |
392 |
161 |
25 |
2.85 |
4.00 |
10.318 |
0.846 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]