LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_254.5wLII_11212_105
Total number of 3D structures: 121
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
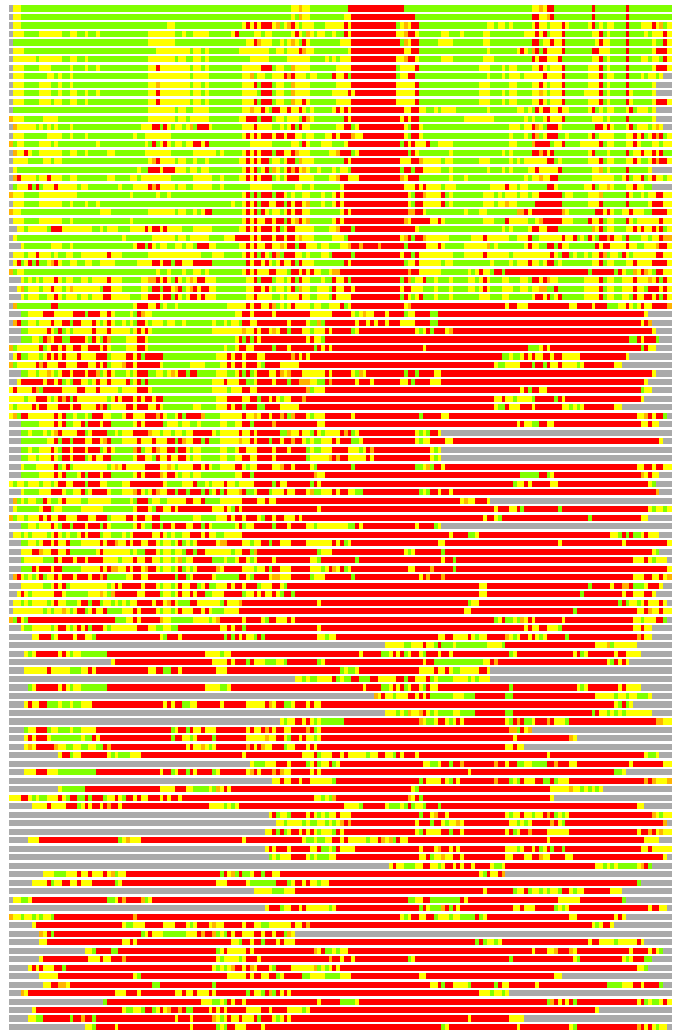
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2ho3_D |
304 |
176 |
156 |
1.15 |
19.87 |
85.928 |
12.508 |
T P |
| 2ho5_A |
305 |
176 |
153 |
1.25 |
20.92 |
83.635 |
11.319 |
T P |
| 3e18_A |
348 |
176 |
149 |
1.97 |
4.03 |
69.713 |
7.205 |
T P |
| 3evn_A |
316 |
176 |
152 |
1.98 |
9.87 |
68.290 |
7.323 |
T P |
| 2glx_A |
332 |
176 |
151 |
1.83 |
4.64 |
66.654 |
7.811 |
T P |
| 1ydw_B |
350 |
176 |
148 |
1.89 |
9.46 |
65.568 |
7.427 |
T P |
| 3e9m_A |
321 |
176 |
154 |
2.08 |
8.44 |
65.188 |
7.074 |
T P |
| 1ofg_A |
381 |
176 |
151 |
2.12 |
5.96 |
64.282 |
6.811 |
T P |
| 2o4u_X |
331 |
176 |
151 |
2.08 |
7.28 |
64.129 |
6.935 |
T P |
| 1h6d_A |
383 |
176 |
150 |
2.11 |
5.33 |
63.556 |
6.797 |
T P |
| 1evj_A |
340 |
176 |
151 |
2.15 |
5.30 |
62.615 |
6.712 |
T P |
| 1ryd_A |
381 |
176 |
149 |
2.11 |
5.37 |
62.563 |
6.757 |
T P |
| 3c1a_A |
307 |
176 |
142 |
2.00 |
6.34 |
61.792 |
6.751 |
T P |
| 3dty_A |
374 |
176 |
145 |
2.02 |
8.97 |
61.714 |
6.846 |
T P |
| 3db2_A |
347 |
176 |
140 |
1.98 |
9.29 |
60.165 |
6.745 |
T P |
| 3e82_D |
350 |
176 |
140 |
2.09 |
10.00 |
60.092 |
6.397 |
T P |
| 1zh8_A |
325 |
176 |
146 |
2.05 |
10.96 |
59.758 |
6.804 |
T P |
| 3f4l_A |
344 |
176 |
140 |
2.10 |
10.71 |
59.515 |
6.374 |
T P |
| 3ezy_A |
334 |
176 |
145 |
2.18 |
5.52 |
59.489 |
6.364 |
T P |
| 3cea_A |
342 |
176 |
139 |
1.99 |
5.76 |
59.379 |
6.657 |
T P |
| 3fd8_D |
349 |
176 |
144 |
2.29 |
9.03 |
58.561 |
6.036 |
T P |
| 1tlt_B |
305 |
176 |
142 |
2.30 |
9.15 |
58.077 |
5.929 |
T P |
| 1gcu_A |
292 |
176 |
139 |
2.12 |
7.91 |
57.258 |
6.249 |
T P |
| 2h63_D |
285 |
176 |
133 |
2.09 |
9.02 |
57.231 |
6.081 |
T P |
| 3ec7_A |
336 |
176 |
133 |
2.18 |
6.02 |
56.839 |
5.826 |
T P |
| 1lc0_A |
290 |
176 |
134 |
2.18 |
8.21 |
56.178 |
5.883 |
T P |
| 3fhl_C |
338 |
176 |
130 |
2.21 |
9.23 |
53.960 |
5.629 |
T P |
| 2p2s_A |
333 |
176 |
136 |
2.30 |
2.21 |
53.326 |
5.658 |
T P |
| 2ixa_A |
426 |
176 |
131 |
2.34 |
6.11 |
51.316 |
5.359 |
T P |
| 3btu_A |
392 |
176 |
138 |
2.51 |
6.52 |
50.639 |
5.278 |
T P |
| 3euw_A |
333 |
176 |
128 |
2.25 |
5.47 |
50.534 |
5.445 |
T P |
| 1xea_A |
311 |
176 |
113 |
2.13 |
5.31 |
50.394 |
5.068 |
T P |
| 2nvw_A |
413 |
176 |
135 |
2.41 |
5.19 |
50.215 |
5.387 |
T P |
| 3btv_B |
392 |
176 |
134 |
2.41 |
3.73 |
50.154 |
5.332 |
T P |
| 3e1k_A |
395 |
176 |
133 |
2.35 |
5.26 |
50.082 |
5.436 |
T P |
| 1ff9_A |
447 |
176 |
98 |
2.27 |
8.16 |
40.805 |
4.140 |
T P |
| 2gb3_B |
393 |
176 |
69 |
2.30 |
7.25 |
29.028 |
2.879 |
T P |
| 1xi9_C |
393 |
176 |
71 |
2.55 |
9.86 |
28.576 |
2.682 |
T P |
| 1v2d_A |
365 |
176 |
77 |
2.61 |
12.99 |
28.465 |
2.838 |
T P |
| 1svv_B |
342 |
176 |
58 |
1.98 |
5.17 |
26.822 |
2.786 |
T P |
| 1o4s_B |
384 |
176 |
65 |
2.34 |
9.23 |
26.702 |
2.668 |
T P |
| 1i29_A |
405 |
176 |
64 |
2.53 |
12.50 |
25.864 |
2.433 |
T P |
| 1c0n_A |
404 |
176 |
69 |
2.81 |
14.49 |
25.789 |
2.374 |
T P |
| 1u08_A |
382 |
176 |
67 |
2.69 |
7.46 |
25.416 |
2.401 |
T P |
| 1j32_A |
388 |
176 |
64 |
2.56 |
6.25 |
25.343 |
2.403 |
T P |
| 3dyd_A |
388 |
176 |
63 |
2.61 |
7.94 |
24.925 |
2.321 |
T P |
| 3cai_A |
396 |
176 |
65 |
2.62 |
12.31 |
24.845 |
2.387 |
T P |
| 1jf9_A |
405 |
176 |
66 |
2.67 |
13.64 |
24.746 |
2.383 |
T P |
| 2hdy_A |
403 |
176 |
66 |
2.80 |
10.61 |
24.619 |
2.275 |
T P |
| 3dr4_C |
368 |
176 |
60 |
2.36 |
10.00 |
24.465 |
2.436 |
T P |
| 1bw0_B |
412 |
176 |
67 |
2.72 |
2.99 |
24.394 |
2.377 |
T P |
| 3b46_B |
424 |
176 |
67 |
2.66 |
7.46 |
24.305 |
2.426 |
T P |
| 2r5e_A |
419 |
176 |
63 |
2.39 |
7.94 |
24.214 |
2.530 |
T P |
| 1yiz_A |
417 |
176 |
64 |
2.59 |
9.38 |
23.743 |
2.379 |
T P |
| 1p3w_B |
385 |
176 |
69 |
2.88 |
2.90 |
23.742 |
2.313 |
T P |
| 3bn1_A |
367 |
176 |
59 |
2.35 |
10.17 |
23.591 |
2.410 |
T P |
| 2gms_A |
390 |
176 |
59 |
2.67 |
8.47 |
23.531 |
2.131 |
T P |
| 2zc0_A |
405 |
176 |
66 |
2.66 |
10.61 |
23.184 |
2.394 |
T P |
| 1qvr_A |
803 |
176 |
62 |
2.51 |
11.29 |
23.171 |
2.373 |
T P |
| 3fd0_A |
407 |
176 |
54 |
2.25 |
7.41 |
22.838 |
2.297 |
T P |
| 1kmj_A |
405 |
176 |
57 |
2.65 |
12.28 |
22.680 |
2.070 |
T P |
| 2o0r_A |
384 |
176 |
59 |
2.44 |
15.25 |
22.443 |
2.326 |
T P |
| 1n2t_A |
385 |
176 |
60 |
2.55 |
5.00 |
21.881 |
2.263 |
T P |
| 1y4i_A |
396 |
176 |
61 |
2.88 |
8.20 |
21.869 |
2.048 |
T P |
| 3b8x_B |
388 |
176 |
58 |
2.58 |
12.07 |
21.856 |
2.167 |
T P |
| 2fnu_B |
374 |
176 |
58 |
2.61 |
18.97 |
21.634 |
2.138 |
T P |
| 2rfv_A |
390 |
176 |
59 |
2.88 |
6.78 |
21.510 |
1.983 |
T P |
| 3dr7_A |
367 |
176 |
55 |
2.71 |
9.09 |
21.489 |
1.955 |
T P |
| 1eg5_B |
365 |
176 |
59 |
2.82 |
8.47 |
21.272 |
2.023 |
T P |
| 1ecx_B |
365 |
176 |
55 |
2.86 |
7.27 |
21.207 |
1.859 |
T P |
| 1elu_A |
382 |
176 |
60 |
2.57 |
5.00 |
21.065 |
2.251 |
T P |
| 2r0t_A |
386 |
176 |
57 |
2.78 |
10.53 |
20.532 |
1.979 |
T P |
| 1t3i_A |
406 |
176 |
50 |
2.55 |
12.00 |
20.121 |
1.885 |
T P |
| 2z61_A |
368 |
176 |
48 |
2.61 |
2.08 |
18.186 |
1.774 |
T P |
| 1fx7_A |
230 |
176 |
45 |
2.70 |
4.44 |
17.681 |
1.607 |
T P |
| 2fbi_A |
136 |
176 |
41 |
2.40 |
7.32 |
16.868 |
1.642 |
T P |
| 2z3y_A |
643 |
176 |
43 |
2.59 |
4.65 |
16.817 |
1.596 |
T P |
| 3cta_A |
187 |
176 |
39 |
2.43 |
5.13 |
16.258 |
1.542 |
T P |
| 2fxa_B |
180 |
176 |
45 |
2.79 |
0.00 |
16.103 |
1.556 |
T P |
| 3caz_B |
210 |
176 |
42 |
2.44 |
4.76 |
16.074 |
1.655 |
T P |
| 2h94_A |
647 |
176 |
41 |
2.56 |
2.44 |
16.015 |
1.539 |
T P |
| 3fm5_B |
140 |
176 |
39 |
2.43 |
2.56 |
15.524 |
1.542 |
T P |
| 3cjn_A |
146 |
176 |
39 |
2.74 |
2.56 |
15.332 |
1.375 |
T P |
| 3bpv_A |
137 |
176 |
40 |
2.53 |
5.00 |
15.153 |
1.524 |
T P |
| 3bro_B |
140 |
176 |
38 |
2.66 |
5.26 |
15.115 |
1.377 |
T P |
| 2fa5_B |
142 |
176 |
39 |
2.79 |
7.69 |
15.052 |
1.348 |
T P |
| 3deu_A |
132 |
176 |
39 |
2.66 |
5.13 |
15.049 |
1.415 |
T P |
| 3bj6_A |
152 |
176 |
38 |
2.96 |
5.26 |
14.884 |
1.240 |
T P |
| 2dw4_A |
634 |
176 |
40 |
2.65 |
0.00 |
14.849 |
1.454 |
T P |
| 2fbh_A |
137 |
176 |
38 |
2.61 |
7.89 |
14.836 |
1.405 |
T P |
| 3e6m_D |
149 |
176 |
36 |
2.48 |
11.11 |
14.698 |
1.396 |
T P |
| 3bja_A |
139 |
176 |
39 |
2.93 |
10.26 |
14.674 |
1.287 |
T P |
| 1z91_A |
137 |
176 |
38 |
2.50 |
2.63 |
14.439 |
1.460 |
T P |
| 2hko_A |
647 |
176 |
37 |
2.73 |
8.11 |
14.243 |
1.307 |
T P |
| 2isz_A |
140 |
176 |
40 |
2.76 |
5.00 |
14.227 |
1.397 |
T P |
| 3eco_A |
135 |
176 |
38 |
2.77 |
2.63 |
14.089 |
1.323 |
T P |
| 3boq_B |
138 |
176 |
37 |
2.81 |
0.00 |
14.000 |
1.274 |
T P |
| 2nnn_A |
134 |
176 |
38 |
3.00 |
5.26 |
13.695 |
1.224 |
T P |
| 2iw5_A |
666 |
176 |
38 |
2.92 |
5.26 |
13.436 |
1.260 |
T P |
| 2fbk_A |
156 |
176 |
35 |
2.89 |
8.57 |
13.224 |
1.170 |
T P |
| 2bv6_A |
137 |
176 |
33 |
2.78 |
15.15 |
13.214 |
1.145 |
T P |
| 2hr3_D |
140 |
176 |
33 |
2.45 |
9.09 |
13.209 |
1.295 |
T P |
| 2eth_A |
141 |
176 |
34 |
2.85 |
0.00 |
13.172 |
1.151 |
T P |
| 1s3j_A |
143 |
176 |
34 |
2.61 |
8.82 |
13.091 |
1.252 |
T P |
| 1lnw_A |
141 |
176 |
31 |
2.63 |
6.45 |
12.928 |
1.134 |
T P |
| 2a61_A |
142 |
176 |
32 |
2.47 |
9.38 |
12.887 |
1.246 |
T P |
| 2pex_B |
137 |
176 |
33 |
2.72 |
15.15 |
12.823 |
1.170 |
T P |
| 1jgs_A |
138 |
176 |
35 |
2.64 |
5.71 |
12.815 |
1.276 |
T P |
| 2rdp_A |
140 |
176 |
35 |
2.85 |
0.00 |
12.801 |
1.187 |
T P |
| 3ech_B |
135 |
176 |
31 |
2.66 |
3.23 |
12.697 |
1.122 |
T P |
| 1lj9_A |
144 |
176 |
32 |
2.88 |
9.38 |
12.297 |
1.074 |
T P |
| 2pfb_A |
129 |
176 |
31 |
2.88 |
19.35 |
12.078 |
1.042 |
T P |
| 1p4x_A |
250 |
176 |
29 |
2.88 |
3.45 |
11.948 |
0.973 |
T P |
| 1f5n_A |
570 |
176 |
32 |
2.92 |
6.25 |
11.800 |
1.060 |
T P |
| 3cdh_B |
136 |
176 |
34 |
2.71 |
14.71 |
11.774 |
1.211 |
T P |
| 2v1d_A |
666 |
176 |
29 |
2.51 |
0.00 |
11.773 |
1.110 |
T P |
| 3bpx_A |
147 |
176 |
30 |
2.79 |
6.67 |
11.684 |
1.037 |
T P |
| 3bdd_A |
140 |
176 |
28 |
2.43 |
10.71 |
11.503 |
1.109 |
T P |
| 2qww_A |
146 |
176 |
29 |
2.81 |
3.45 |
11.436 |
0.995 |
T P |
| 2gxg_A |
140 |
176 |
28 |
2.69 |
7.14 |
11.322 |
1.005 |
T P |
| 2nyx_B |
147 |
176 |
23 |
2.76 |
4.35 |
9.765 |
0.804 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]