LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_258.5wLII_11212_125
Total number of 3D structures: 58
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
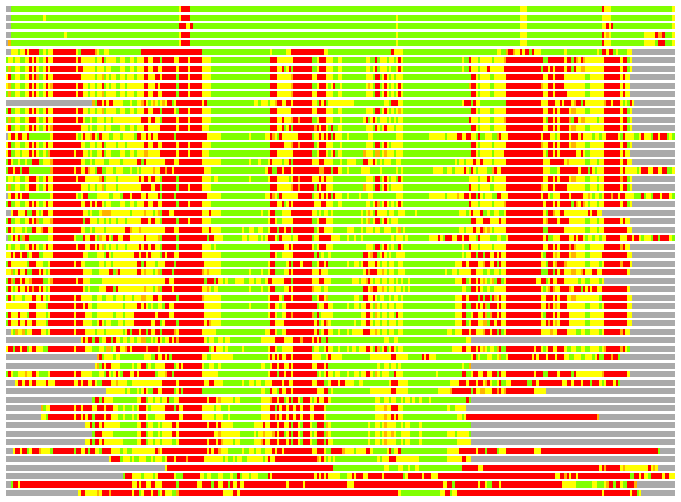
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1oq4_A |
346 |
286 |
279 |
0.80 |
17.56 |
96.556 |
31.115 |
T P |
| 2j2f_A |
348 |
286 |
280 |
0.89 |
17.14 |
96.198 |
28.240 |
T P |
| 1afr_A |
345 |
286 |
278 |
0.84 |
17.27 |
95.944 |
29.534 |
T P |
| 2uw1_B |
338 |
286 |
277 |
1.07 |
16.61 |
94.786 |
23.607 |
T P |
| 2uw1_A |
327 |
286 |
277 |
1.02 |
16.61 |
94.709 |
24.655 |
T P |
| 1za0_A |
242 |
286 |
194 |
1.62 |
19.07 |
63.610 |
11.303 |
T P |
| 2av8_A |
340 |
286 |
181 |
2.28 |
12.15 |
44.874 |
7.604 |
T P |
| 1yfd_B |
341 |
286 |
178 |
2.28 |
13.48 |
44.594 |
7.476 |
T P |
| 1pj0_A |
340 |
286 |
182 |
2.33 |
12.64 |
43.768 |
7.492 |
T P |
| 1piy_A |
340 |
286 |
182 |
2.35 |
10.99 |
43.571 |
7.425 |
T P |
| 1piz_A |
340 |
286 |
181 |
2.34 |
11.60 |
43.058 |
7.406 |
T P |
| 2oc5_A |
222 |
286 |
160 |
2.23 |
12.50 |
42.599 |
6.863 |
T P |
| 1pm2_A |
339 |
286 |
180 |
2.31 |
11.67 |
42.506 |
7.475 |
T P |
| 1pfr_A |
340 |
286 |
181 |
2.33 |
13.26 |
42.441 |
7.437 |
T P |
| 1mxr_A |
339 |
286 |
180 |
2.30 |
11.11 |
42.391 |
7.485 |
T P |
| 1t0q_A |
491 |
286 |
188 |
2.44 |
11.17 |
42.375 |
7.394 |
T P |
| 1rib_A |
340 |
286 |
179 |
2.35 |
11.73 |
42.265 |
7.295 |
T P |
| 1biq_A |
339 |
286 |
181 |
2.33 |
11.05 |
42.210 |
7.460 |
T P |
| 2ani_A |
317 |
286 |
187 |
2.42 |
12.83 |
42.188 |
7.409 |
T P |
| 3dhi_A |
498 |
286 |
177 |
2.35 |
12.43 |
42.170 |
7.219 |
T P |
| 1av8_A |
340 |
286 |
180 |
2.41 |
11.11 |
42.059 |
7.158 |
T P |
| 1rnr_A |
339 |
286 |
182 |
2.39 |
11.54 |
41.859 |
7.307 |
T P |
| 2rdb_A |
491 |
286 |
182 |
2.36 |
10.99 |
41.837 |
7.405 |
T P |
| 1biq_B |
341 |
286 |
172 |
2.32 |
9.88 |
41.794 |
7.112 |
T P |
| 1uzr_B |
288 |
286 |
180 |
2.55 |
11.67 |
41.792 |
6.797 |
T P |
| 2alx_A |
339 |
286 |
175 |
2.32 |
9.71 |
41.249 |
7.233 |
T P |
| 1syy_A |
317 |
286 |
181 |
2.46 |
12.71 |
40.835 |
7.060 |
T P |
| 2inc_A |
491 |
286 |
179 |
2.42 |
12.29 |
40.746 |
7.093 |
T P |
| 1rsr_B |
341 |
286 |
172 |
2.38 |
11.63 |
40.677 |
6.930 |
T P |
| 2rcc_A |
313 |
286 |
169 |
2.34 |
13.02 |
40.637 |
6.931 |
T P |
| 1h0o_A |
288 |
286 |
177 |
2.38 |
11.30 |
40.443 |
7.125 |
T P |
| 1r2f_A |
283 |
286 |
174 |
2.54 |
11.49 |
40.387 |
6.585 |
T P |
| 2iyh_A |
275 |
286 |
174 |
2.45 |
11.49 |
40.282 |
6.823 |
T P |
| 3dhz_B |
296 |
286 |
170 |
2.43 |
9.41 |
40.093 |
6.725 |
T P |
| 2uw2_A |
275 |
286 |
170 |
2.40 |
12.94 |
39.589 |
6.798 |
T P |
| 1jk0_A |
334 |
286 |
173 |
2.45 |
8.67 |
39.353 |
6.783 |
T P |
| 2o1z_A |
288 |
286 |
165 |
2.38 |
13.94 |
38.991 |
6.643 |
T P |
| 1smq_A |
329 |
286 |
163 |
2.38 |
9.20 |
38.422 |
6.561 |
T P |
| 2p1i_A |
258 |
286 |
166 |
2.45 |
13.25 |
38.270 |
6.499 |
T P |
| 3fvb_A |
161 |
286 |
131 |
2.03 |
18.32 |
37.907 |
6.151 |
T P |
| 2vux_A |
272 |
286 |
161 |
2.45 |
10.56 |
37.873 |
6.304 |
T P |
| 3ez0_A |
216 |
286 |
162 |
2.22 |
9.88 |
37.574 |
6.971 |
T P |
| 1nfv_B |
170 |
286 |
126 |
1.86 |
17.46 |
37.255 |
6.436 |
T P |
| 1jk0_B |
265 |
286 |
160 |
2.33 |
7.50 |
36.524 |
6.576 |
T P |
| 2jcd_A |
315 |
286 |
148 |
2.52 |
10.14 |
34.539 |
5.658 |
T P |
| 2itb_A |
199 |
286 |
129 |
2.24 |
15.50 |
33.183 |
5.506 |
T P |
| 1eum_A |
161 |
286 |
127 |
2.13 |
15.75 |
32.036 |
5.688 |
T P |
| 2v8t_A |
302 |
286 |
125 |
2.10 |
10.40 |
31.813 |
5.673 |
T P |
| 2cwl_A |
299 |
286 |
126 |
2.17 |
11.11 |
31.518 |
5.550 |
T P |
| 1vlg_A |
164 |
286 |
126 |
2.23 |
14.29 |
31.464 |
5.417 |
T P |
| 2jd6_0 |
167 |
286 |
128 |
2.33 |
15.62 |
31.320 |
5.258 |
T P |
| 1z4a_A |
164 |
286 |
126 |
2.28 |
13.49 |
30.870 |
5.283 |
T P |
| 3cht_A |
301 |
286 |
126 |
2.46 |
11.11 |
29.612 |
4.928 |
T P |
| 2oh3_A |
149 |
286 |
112 |
2.40 |
12.50 |
27.862 |
4.482 |
T P |
| 1j30_A |
141 |
286 |
75 |
2.07 |
13.33 |
22.992 |
3.461 |
T P |
| 3ezu_A |
336 |
286 |
82 |
2.61 |
4.88 |
19.119 |
3.028 |
T P |
| 1j0a_A |
325 |
286 |
59 |
2.72 |
8.47 |
13.319 |
2.089 |
T P |
| 2e0z_A |
236 |
286 |
53 |
2.46 |
9.43 |
13.152 |
2.071 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]