LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_26.5wLII_10665_6
Total number of 3D structures: 40
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
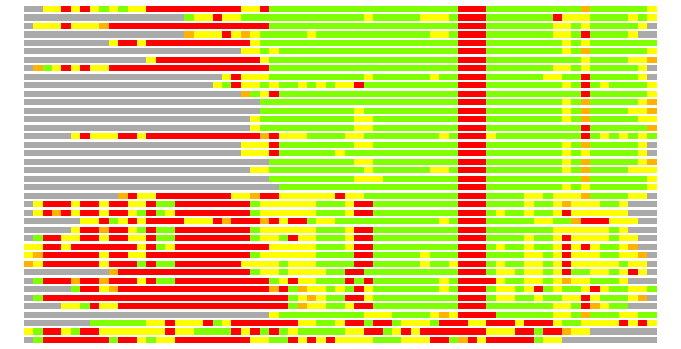
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2jzx_A |
160 |
67 |
49 |
1.70 |
22.45 |
67.045 |
2.715 |
T P |
| 1x4m_A |
94 |
67 |
45 |
1.84 |
24.44 |
60.120 |
2.324 |
T P |
| 2anr_A |
155 |
67 |
44 |
1.83 |
20.45 |
59.155 |
2.277 |
T P |
| 2dgr_A |
83 |
67 |
43 |
2.00 |
16.28 |
58.168 |
2.048 |
T P |
| 1we8_A |
104 |
67 |
42 |
1.62 |
28.57 |
58.097 |
2.446 |
T P |
| 1wvn_A |
74 |
67 |
41 |
1.63 |
19.51 |
57.443 |
2.366 |
T P |
| 2axy_A |
72 |
67 |
40 |
1.57 |
20.00 |
57.260 |
2.396 |
T P |
| 2ann_A |
148 |
67 |
43 |
1.72 |
18.60 |
56.685 |
2.363 |
T P |
| 1khm_A |
89 |
67 |
40 |
1.64 |
12.50 |
56.533 |
2.293 |
T P |
| 2qnd_B |
143 |
67 |
41 |
1.92 |
19.51 |
55.735 |
2.034 |
T P |
| 1j5k_A |
76 |
67 |
39 |
1.58 |
15.38 |
55.689 |
2.315 |
T P |
| 1ztg_B |
74 |
67 |
39 |
1.66 |
23.08 |
55.610 |
2.221 |
T P |
| 2p2r_A |
73 |
67 |
39 |
1.56 |
20.51 |
55.479 |
2.356 |
T P |
| 1dtj_A |
74 |
67 |
40 |
1.63 |
20.00 |
55.355 |
2.314 |
T P |
| 1dt4_A |
73 |
67 |
39 |
1.52 |
20.51 |
55.080 |
2.403 |
T P |
| 2opu_A |
89 |
67 |
42 |
2.08 |
21.43 |
55.026 |
1.928 |
T P |
| 1zzk_A |
80 |
67 |
39 |
1.68 |
17.95 |
54.451 |
2.187 |
T P |
| 1x4n_A |
92 |
67 |
39 |
1.71 |
25.64 |
54.318 |
2.151 |
T P |
| 1ec6_A |
87 |
67 |
38 |
1.61 |
21.05 |
53.895 |
2.226 |
T P |
| 2opv_A |
85 |
67 |
40 |
1.85 |
27.50 |
53.576 |
2.048 |
T P |
| 1j4w_A |
145 |
67 |
38 |
1.55 |
21.05 |
52.625 |
2.309 |
T P |
| 2hh3_A |
71 |
67 |
37 |
1.42 |
32.43 |
52.173 |
2.438 |
T P |
| 2ctk_A |
104 |
67 |
41 |
2.34 |
17.07 |
49.872 |
1.677 |
T P |
| 1i94_C |
206 |
67 |
42 |
2.32 |
30.95 |
49.800 |
1.734 |
T P |
| 2gy9_C |
206 |
67 |
42 |
2.35 |
19.05 |
49.690 |
1.716 |
T P |
| 1wf3_A |
296 |
67 |
40 |
2.32 |
17.50 |
49.165 |
1.655 |
T P |
| 2zkq_c |
192 |
67 |
41 |
2.24 |
19.51 |
48.334 |
1.751 |
T P |
| 3fic_C |
206 |
67 |
42 |
2.28 |
30.95 |
48.245 |
1.765 |
T P |
| 2vqe_C |
206 |
67 |
38 |
2.19 |
34.21 |
47.363 |
1.661 |
T P |
| 1pns_C |
206 |
67 |
41 |
2.53 |
34.15 |
47.306 |
1.557 |
T P |
| 3bbn_C |
217 |
67 |
43 |
2.34 |
4.65 |
47.225 |
1.762 |
T P |
| 2pt7_G |
145 |
67 |
36 |
2.21 |
19.44 |
45.571 |
1.558 |
T P |
| 2qal_C |
206 |
67 |
37 |
2.29 |
18.92 |
44.801 |
1.549 |
T P |
| 2cxc_A |
138 |
67 |
37 |
2.38 |
24.32 |
44.068 |
1.492 |
T P |
| 1vs5_C |
206 |
67 |
33 |
2.09 |
21.21 |
43.181 |
1.507 |
T P |
| 1wh9_A |
92 |
67 |
35 |
2.18 |
14.29 |
43.115 |
1.536 |
T P |
| 2hh2_A |
79 |
67 |
37 |
2.06 |
24.32 |
41.902 |
1.714 |
T P |
| 3cdi_A |
516 |
67 |
36 |
2.32 |
5.56 |
38.113 |
1.490 |
T P |
| 1e3p_A |
645 |
67 |
38 |
2.44 |
13.16 |
37.804 |
1.494 |
T P |
| 1e3h_A |
601 |
67 |
28 |
2.45 |
3.57 |
31.683 |
1.097 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]