LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_264.5wLII_11226_11
Total number of 3D structures: 79
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
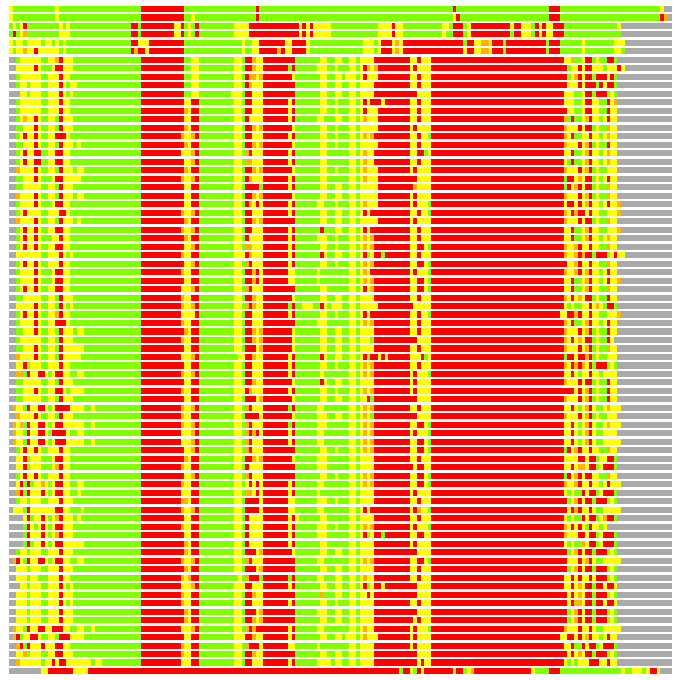
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1qsa_A |
618 |
185 |
166 |
0.46 |
15.66 |
89.654 |
29.604 |
T P |
| 1sly_A |
618 |
185 |
166 |
0.70 |
15.66 |
89.599 |
20.797 |
T P |
| 1gbs_A |
185 |
185 |
119 |
1.88 |
10.92 |
50.496 |
6.016 |
T P |
| 153l_A |
185 |
185 |
119 |
1.86 |
9.24 |
49.056 |
6.057 |
T P |
| 1ltm_A |
309 |
185 |
110 |
2.13 |
11.82 |
44.159 |
4.927 |
T P |
| 1qus_A |
312 |
185 |
109 |
2.19 |
11.93 |
43.065 |
4.759 |
T P |
| 1flu_A |
129 |
185 |
92 |
2.16 |
9.78 |
39.023 |
4.079 |
T P |
| 1lsg_A |
144 |
185 |
92 |
2.29 |
7.61 |
38.971 |
3.851 |
T P |
| 1at5_A |
127 |
185 |
90 |
2.19 |
10.00 |
38.633 |
3.931 |
T P |
| 1iot_A |
129 |
185 |
89 |
2.23 |
10.11 |
37.881 |
3.816 |
T P |
| 1heq_A |
129 |
185 |
89 |
2.14 |
10.11 |
37.708 |
3.981 |
T P |
| 1gbx_A |
130 |
185 |
91 |
2.19 |
7.69 |
37.214 |
3.971 |
T P |
| 1di3_A |
130 |
185 |
91 |
2.18 |
7.69 |
37.053 |
3.983 |
T P |
| 2lhm_A |
130 |
185 |
93 |
2.30 |
8.60 |
36.806 |
3.867 |
T P |
| 1ir9_A |
129 |
185 |
92 |
2.31 |
6.52 |
36.735 |
3.822 |
T P |
| 1gfu_A |
130 |
185 |
91 |
2.27 |
8.79 |
36.709 |
3.833 |
T P |
| 1flw_A |
129 |
185 |
92 |
2.31 |
7.61 |
36.702 |
3.812 |
T P |
| 1ip2_A |
130 |
185 |
91 |
2.28 |
7.69 |
36.666 |
3.824 |
T P |
| 1ouh_A |
130 |
185 |
92 |
2.30 |
7.61 |
36.621 |
3.826 |
T P |
| 1lzd_A |
129 |
185 |
93 |
2.40 |
7.53 |
36.580 |
3.720 |
T P |
| 2vb1_A |
129 |
185 |
91 |
2.30 |
7.69 |
36.579 |
3.796 |
T P |
| 1flq_A |
129 |
185 |
90 |
2.21 |
8.89 |
36.544 |
3.894 |
T P |
| 1ioq_A |
129 |
185 |
93 |
2.38 |
7.53 |
36.513 |
3.745 |
T P |
| 1i20_A |
130 |
185 |
90 |
2.29 |
7.78 |
36.484 |
3.773 |
T P |
| 1gb6_A |
130 |
185 |
90 |
2.33 |
7.78 |
36.414 |
3.709 |
T P |
| 1ior_A |
129 |
185 |
91 |
2.33 |
7.69 |
36.413 |
3.743 |
T P |
| 1lhj_A |
130 |
185 |
90 |
2.25 |
7.78 |
36.384 |
3.822 |
T P |
| 1i22_A |
130 |
185 |
92 |
2.27 |
6.52 |
36.370 |
3.876 |
T P |
| 1gb7_A |
130 |
185 |
92 |
2.37 |
8.70 |
36.367 |
3.729 |
T P |
| 1nbz_C |
129 |
185 |
91 |
2.27 |
9.89 |
36.332 |
3.836 |
T P |
| 1gb9_A |
130 |
185 |
91 |
2.34 |
7.69 |
36.330 |
3.725 |
T P |
| 1yan_A |
130 |
185 |
90 |
2.31 |
8.89 |
36.313 |
3.741 |
T P |
| 1cj6_A |
130 |
185 |
90 |
2.25 |
7.78 |
36.306 |
3.828 |
T P |
| 1ge3_A |
130 |
185 |
90 |
2.26 |
7.78 |
36.263 |
3.816 |
T P |
| 1lsn_A |
129 |
185 |
92 |
2.35 |
9.78 |
36.259 |
3.753 |
T P |
| 1hhl_A |
129 |
185 |
94 |
2.38 |
8.51 |
36.229 |
3.798 |
T P |
| 2nwd_X |
130 |
185 |
91 |
2.31 |
7.69 |
36.192 |
3.773 |
T P |
| 1wqm_A |
130 |
185 |
89 |
2.26 |
7.87 |
36.177 |
3.768 |
T P |
| 1ios_A |
129 |
185 |
91 |
2.33 |
7.69 |
36.165 |
3.743 |
T P |
| 1fly_A |
129 |
185 |
90 |
2.30 |
10.00 |
36.164 |
3.744 |
T P |
| 1kxw_A |
129 |
185 |
91 |
2.34 |
7.69 |
36.123 |
3.736 |
T P |
| 2ihl_A |
129 |
185 |
90 |
2.31 |
7.78 |
36.089 |
3.732 |
T P |
| 1eqe_A |
130 |
185 |
88 |
2.24 |
6.82 |
35.992 |
3.756 |
T P |
| 1gft_A |
130 |
185 |
91 |
2.33 |
5.49 |
35.971 |
3.747 |
T P |
| 1lsm_A |
129 |
185 |
90 |
2.32 |
8.89 |
35.947 |
3.725 |
T P |
| 2iff_Y |
129 |
185 |
89 |
2.33 |
7.87 |
35.906 |
3.668 |
T P |
| 1at6_A |
128 |
185 |
89 |
2.27 |
8.99 |
35.868 |
3.761 |
T P |
| 1i1z_A |
130 |
185 |
92 |
2.38 |
5.43 |
35.830 |
3.703 |
T P |
| 1heo_A |
129 |
185 |
90 |
2.32 |
7.78 |
35.809 |
3.727 |
T P |
| 1lhl_A |
130 |
185 |
93 |
2.39 |
5.38 |
35.781 |
3.741 |
T P |
| 2meb_A |
130 |
185 |
90 |
2.37 |
6.67 |
35.775 |
3.646 |
T P |
| 1cj7_A |
130 |
185 |
92 |
2.38 |
5.43 |
35.743 |
3.716 |
T P |
| 1gaz_A |
130 |
185 |
90 |
2.35 |
7.78 |
35.728 |
3.670 |
T P |
| 1c46_A |
131 |
185 |
87 |
2.25 |
5.75 |
35.669 |
3.698 |
T P |
| 1ouf_A |
130 |
185 |
87 |
2.26 |
5.75 |
35.624 |
3.693 |
T P |
| 1gfh_A |
130 |
185 |
89 |
2.31 |
7.87 |
35.611 |
3.699 |
T P |
| 1gb5_A |
130 |
185 |
90 |
2.37 |
5.56 |
35.541 |
3.650 |
T P |
| 1ckd_A |
130 |
185 |
87 |
2.26 |
5.75 |
35.537 |
3.689 |
T P |
| 1lsy_A |
129 |
185 |
88 |
2.23 |
9.09 |
35.420 |
3.777 |
T P |
| 1gb8_A |
130 |
185 |
90 |
2.38 |
6.67 |
35.416 |
3.627 |
T P |
| 1ioc_A |
133 |
185 |
87 |
2.19 |
4.60 |
35.388 |
3.800 |
T P |
| 134l_A |
130 |
185 |
86 |
2.31 |
5.81 |
35.289 |
3.573 |
T P |
| 1tdy_A |
130 |
185 |
84 |
2.19 |
5.95 |
35.223 |
3.668 |
T P |
| 1c7p_A |
133 |
185 |
86 |
2.21 |
4.65 |
35.162 |
3.720 |
T P |
| 1ir7_A |
129 |
185 |
86 |
2.20 |
9.30 |
35.145 |
3.732 |
T P |
| 1ouc_A |
130 |
185 |
90 |
2.42 |
5.56 |
35.049 |
3.578 |
T P |
| 1hen_A |
129 |
185 |
86 |
2.23 |
9.30 |
35.005 |
3.692 |
T P |
| 1fbi_X |
129 |
185 |
89 |
2.28 |
10.11 |
34.951 |
3.744 |
T P |
| 1nby_C |
129 |
185 |
88 |
2.30 |
11.36 |
34.932 |
3.668 |
T P |
| 3b72_A |
129 |
185 |
86 |
2.27 |
10.47 |
34.864 |
3.621 |
T P |
| 1ndg_C |
129 |
185 |
88 |
2.32 |
10.23 |
34.829 |
3.642 |
T P |
| 1hem_A |
129 |
185 |
85 |
2.22 |
9.41 |
34.752 |
3.662 |
T P |
| 1hep_A |
129 |
185 |
86 |
2.25 |
9.30 |
34.661 |
3.654 |
T P |
| 1wqo_A |
130 |
185 |
91 |
2.46 |
6.59 |
34.632 |
3.555 |
T P |
| 1dkk_A |
129 |
185 |
88 |
2.39 |
9.09 |
34.561 |
3.539 |
T P |
| 2hea_A |
130 |
185 |
85 |
2.20 |
7.06 |
34.292 |
3.692 |
T P |
| 1her_A |
129 |
185 |
86 |
2.26 |
10.47 |
34.281 |
3.648 |
T P |
| 1hml_A |
123 |
185 |
88 |
2.42 |
10.23 |
33.240 |
3.489 |
T P |
| 2ra1_A |
412 |
185 |
45 |
2.19 |
8.89 |
19.124 |
1.962 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]