LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_266.5wLII_11233_28
Total number of 3D structures: 78
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
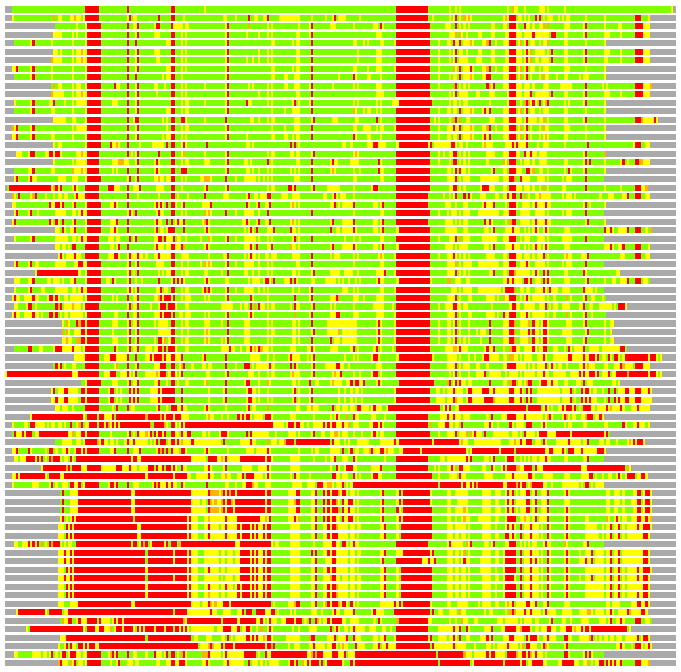
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1l0q_A |
391 |
320 |
291 |
1.02 |
16.49 |
88.273 |
26.028 |
T P |
| 1ri6_A |
333 |
320 |
267 |
1.78 |
9.74 |
75.167 |
14.188 |
T P |
| 3fm0_A |
328 |
320 |
243 |
1.71 |
9.05 |
70.312 |
13.400 |
T P |
| 2gnq_A |
316 |
320 |
245 |
1.72 |
6.53 |
70.218 |
13.448 |
T P |
| 2h9l_A |
321 |
320 |
246 |
1.67 |
10.16 |
70.134 |
13.937 |
T P |
| 2h6n_B |
305 |
320 |
245 |
1.72 |
6.94 |
70.119 |
13.444 |
T P |
| 2g99_A |
304 |
320 |
246 |
1.76 |
6.91 |
70.035 |
13.253 |
T P |
| 2h9m_A |
304 |
320 |
246 |
1.70 |
9.76 |
69.999 |
13.685 |
T P |
| 2h14_A |
303 |
320 |
245 |
1.69 |
10.20 |
69.868 |
13.694 |
T P |
| 1vyh_C |
310 |
320 |
246 |
1.77 |
9.76 |
69.853 |
13.159 |
T P |
| 2cnx_A |
306 |
320 |
246 |
1.76 |
6.10 |
69.778 |
13.198 |
T P |
| 3frx_B |
313 |
320 |
242 |
1.72 |
10.33 |
69.731 |
13.303 |
T P |
| 2g9a_A |
310 |
320 |
246 |
1.70 |
9.76 |
69.718 |
13.667 |
T P |
| 1erj_C |
357 |
320 |
246 |
1.78 |
8.94 |
69.705 |
13.052 |
T P |
| 2co0_A |
304 |
320 |
246 |
1.77 |
8.94 |
69.596 |
13.121 |
T P |
| 3emh_A |
300 |
320 |
245 |
1.71 |
9.80 |
69.560 |
13.505 |
T P |
| 3bws_A |
407 |
320 |
243 |
1.80 |
9.47 |
69.204 |
12.787 |
T P |
| 1gxr_A |
335 |
320 |
239 |
1.66 |
8.79 |
68.938 |
13.548 |
T P |
| 3dm0_A |
675 |
320 |
244 |
1.86 |
9.43 |
68.636 |
12.430 |
T P |
| 2hes_X |
308 |
320 |
239 |
1.74 |
8.37 |
68.085 |
12.969 |
T P |
| 2bcj_B |
339 |
320 |
246 |
1.91 |
9.76 |
67.883 |
12.223 |
T P |
| 1p22_A |
402 |
320 |
233 |
1.76 |
7.73 |
66.647 |
12.500 |
T P |
| 3fgb_A |
349 |
320 |
261 |
1.93 |
13.41 |
66.140 |
12.859 |
T P |
| 2z2n_A |
293 |
320 |
234 |
1.72 |
11.54 |
65.552 |
12.860 |
T P |
| 1tbg_A |
340 |
320 |
244 |
1.88 |
10.66 |
65.473 |
12.309 |
T P |
| 2z2p_A |
293 |
320 |
236 |
1.79 |
11.02 |
65.113 |
12.507 |
T P |
| 2qc5_A |
298 |
320 |
236 |
1.84 |
8.90 |
64.849 |
12.190 |
T P |
| 2pbi_D |
354 |
320 |
244 |
1.86 |
9.02 |
64.445 |
12.478 |
T P |
| 2z2o_C |
299 |
320 |
238 |
1.90 |
9.66 |
63.420 |
11.873 |
T P |
| 3cfv_B |
393 |
320 |
239 |
1.95 |
9.62 |
62.920 |
11.640 |
T P |
| 1got_B |
339 |
320 |
244 |
1.91 |
9.84 |
62.465 |
12.124 |
T P |
| 3bg1_A |
285 |
320 |
222 |
1.86 |
9.01 |
61.850 |
11.319 |
T P |
| 2ece_A |
455 |
320 |
255 |
1.96 |
9.80 |
60.610 |
12.379 |
T P |
| 1a0r_B |
339 |
320 |
241 |
2.02 |
11.20 |
59.953 |
11.344 |
T P |
| 3c99_A |
380 |
320 |
238 |
1.96 |
6.72 |
59.508 |
11.569 |
T P |
| 1nr0_A |
610 |
320 |
245 |
1.98 |
8.16 |
58.557 |
11.785 |
T P |
| 3cfs_B |
383 |
320 |
237 |
1.96 |
7.17 |
58.180 |
11.499 |
T P |
| 2pm6_D |
288 |
320 |
224 |
1.98 |
9.82 |
55.242 |
10.747 |
T P |
| 2pm9_B |
280 |
320 |
223 |
2.02 |
9.87 |
54.698 |
10.534 |
T P |
| 2pm7_D |
288 |
320 |
222 |
1.96 |
9.01 |
54.566 |
10.778 |
T P |
| 1pi6_A |
606 |
320 |
237 |
2.02 |
8.02 |
54.165 |
11.176 |
T P |
| 2ovr_B |
442 |
320 |
211 |
2.19 |
9.48 |
52.337 |
9.208 |
T P |
| 3ei4_B |
368 |
320 |
229 |
2.06 |
6.99 |
52.204 |
10.606 |
T P |
| 1nex_B |
444 |
320 |
214 |
2.24 |
10.28 |
51.985 |
9.144 |
T P |
| 3ewe_A |
255 |
320 |
207 |
1.91 |
10.63 |
49.436 |
10.307 |
T P |
| 2b4e_A |
386 |
320 |
218 |
2.26 |
8.26 |
48.478 |
9.231 |
T P |
| 2aq5_A |
395 |
320 |
218 |
2.30 |
8.26 |
47.060 |
9.072 |
T P |
| 1q7f_B |
282 |
320 |
186 |
2.18 |
9.68 |
44.343 |
8.158 |
T P |
| 2dso_C |
323 |
320 |
177 |
2.26 |
10.17 |
40.602 |
7.498 |
T P |
| 1npe_A |
263 |
320 |
189 |
2.31 |
11.11 |
40.499 |
7.831 |
T P |
| 1r5m_A |
351 |
320 |
193 |
2.32 |
8.29 |
40.314 |
7.969 |
T P |
| 2zkq_a |
306 |
320 |
202 |
2.54 |
11.39 |
39.935 |
7.648 |
T P |
| 2fp8_B |
303 |
320 |
182 |
2.32 |
10.99 |
39.784 |
7.534 |
T P |
| 2dg0_A |
322 |
320 |
175 |
2.17 |
10.29 |
39.240 |
7.716 |
T P |
| 1rwi_A |
256 |
320 |
173 |
2.29 |
8.67 |
39.104 |
7.236 |
T P |
| 1ijq_A |
308 |
320 |
186 |
2.26 |
6.99 |
39.062 |
7.885 |
T P |
| 3dr2_A |
299 |
320 |
175 |
2.30 |
10.29 |
38.692 |
7.294 |
T P |
| 2iau_A |
312 |
320 |
174 |
2.23 |
14.37 |
38.324 |
7.473 |
T P |
| 2iav_A |
312 |
320 |
175 |
2.25 |
14.86 |
37.851 |
7.444 |
T P |
| 2ias_A |
312 |
320 |
172 |
2.21 |
15.12 |
37.785 |
7.432 |
T P |
| 2gvu_A |
312 |
320 |
179 |
2.34 |
13.97 |
37.767 |
7.346 |
T P |
| 2iao_A |
312 |
320 |
185 |
2.36 |
13.51 |
37.677 |
7.532 |
T P |
| 2iap_A |
312 |
320 |
184 |
2.37 |
13.59 |
37.546 |
7.458 |
T P |
| 2dg1_B |
322 |
320 |
166 |
2.10 |
7.83 |
37.343 |
7.530 |
T P |
| 2iax_A |
312 |
320 |
184 |
2.36 |
12.50 |
37.242 |
7.475 |
T P |
| 2iar_A |
312 |
320 |
183 |
2.37 |
12.57 |
37.222 |
7.415 |
T P |
| 2iaq_A |
312 |
320 |
182 |
2.32 |
13.74 |
37.204 |
7.507 |
T P |
| 2gvx_A |
312 |
320 |
185 |
2.37 |
11.89 |
37.162 |
7.478 |
T P |
| 2iat_A |
312 |
320 |
182 |
2.33 |
13.74 |
37.148 |
7.493 |
T P |
| 1pjx_A |
314 |
320 |
183 |
2.33 |
13.66 |
37.125 |
7.530 |
T P |
| 2iaw_A |
312 |
320 |
175 |
2.30 |
13.71 |
36.885 |
7.297 |
T P |
| 1n7d_A |
639 |
320 |
187 |
2.45 |
8.02 |
36.628 |
7.321 |
T P |
| 2qe8_A |
337 |
320 |
174 |
2.31 |
12.64 |
36.561 |
7.206 |
T P |
| 2ghs_A |
295 |
320 |
170 |
2.42 |
11.18 |
35.645 |
6.744 |
T P |
| 2ism_B |
333 |
320 |
177 |
2.39 |
12.99 |
35.624 |
7.099 |
T P |
| 2g8s_A |
348 |
320 |
172 |
2.43 |
9.30 |
34.187 |
6.810 |
T P |
| 3e5z_A |
290 |
320 |
161 |
2.40 |
8.70 |
32.955 |
6.451 |
T P |
| 1v04_A |
332 |
320 |
151 |
2.32 |
14.57 |
31.034 |
6.236 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]