LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_270.5wLII_11233_42
Total number of 3D structures: 85
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
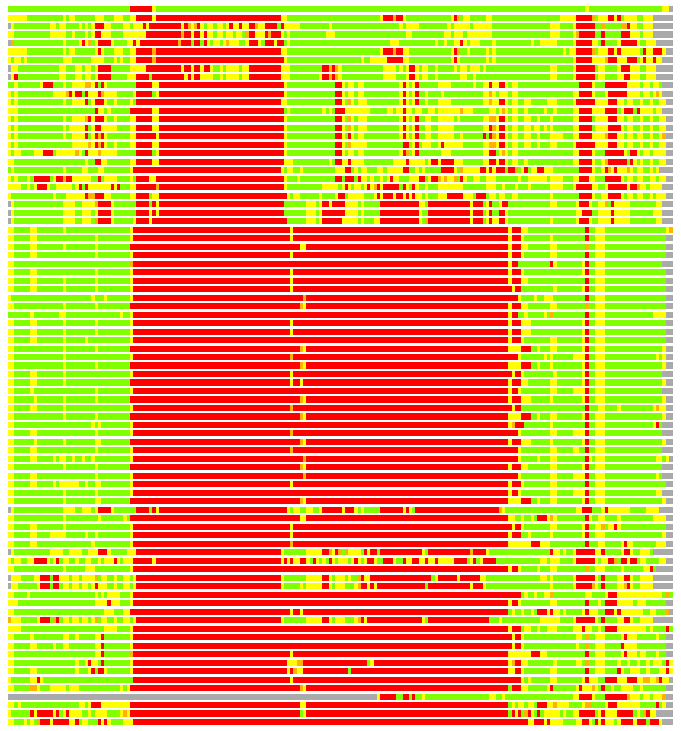
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2in3_A |
204 |
207 |
199 |
0.53 |
18.59 |
95.625 |
31.689 |
T P |
| 3bck_A |
164 |
207 |
143 |
1.92 |
15.38 |
60.169 |
7.070 |
T P |
| 1yzx_A |
218 |
207 |
168 |
2.16 |
14.88 |
59.922 |
7.450 |
T P |
| 1r4w_A |
221 |
207 |
168 |
2.08 |
14.88 |
59.350 |
7.691 |
T P |
| 3fz5_C |
192 |
207 |
157 |
2.00 |
14.01 |
59.095 |
7.464 |
T P |
| 3bci_A |
165 |
207 |
142 |
1.89 |
15.49 |
58.526 |
7.138 |
T P |
| 3bd2_A |
164 |
207 |
140 |
1.89 |
15.71 |
58.317 |
7.050 |
T P |
| 2imf_A |
203 |
207 |
156 |
2.00 |
10.90 |
56.377 |
7.425 |
T P |
| 2imd_A |
203 |
207 |
156 |
2.03 |
10.90 |
56.169 |
7.318 |
T P |
| 1u3a_E |
188 |
207 |
137 |
1.98 |
8.76 |
52.468 |
6.573 |
T P |
| 2b6m_B |
188 |
207 |
141 |
2.05 |
9.22 |
52.088 |
6.551 |
T P |
| 1fvj_A |
188 |
207 |
141 |
2.08 |
8.51 |
51.845 |
6.477 |
T P |
| 2rem_C |
191 |
207 |
141 |
2.08 |
9.93 |
51.812 |
6.472 |
T P |
| 1fvk_A |
188 |
207 |
144 |
2.16 |
9.03 |
51.340 |
6.369 |
T P |
| 1ac1_A |
188 |
207 |
143 |
2.15 |
8.39 |
51.113 |
6.360 |
T P |
| 1bq7_A |
186 |
207 |
141 |
2.10 |
8.51 |
50.770 |
6.396 |
T P |
| 1acv_A |
188 |
207 |
142 |
2.18 |
8.45 |
50.720 |
6.224 |
T P |
| 2b3s_A |
187 |
207 |
139 |
2.12 |
9.35 |
50.218 |
6.271 |
T P |
| 1bed_A |
181 |
207 |
134 |
2.00 |
10.45 |
48.081 |
6.382 |
T P |
| 3c7m_A |
195 |
207 |
132 |
2.11 |
8.33 |
46.148 |
5.974 |
T P |
| 2znm_C |
186 |
207 |
131 |
2.22 |
9.92 |
43.813 |
5.656 |
T P |
| 2ijy_A |
181 |
207 |
129 |
2.30 |
7.75 |
43.145 |
5.365 |
T P |
| 3feu_A |
183 |
207 |
131 |
2.18 |
11.45 |
42.507 |
5.750 |
T P |
| 1jzo_B |
216 |
207 |
114 |
2.05 |
16.67 |
40.743 |
5.299 |
T P |
| 1eej_A |
216 |
207 |
113 |
2.04 |
16.81 |
40.443 |
5.285 |
T P |
| 1jzd_A |
219 |
207 |
112 |
2.11 |
16.96 |
38.802 |
5.057 |
T P |
| 3dxb_D |
216 |
207 |
87 |
1.68 |
13.79 |
38.395 |
4.882 |
T P |
| 2trx_A |
108 |
207 |
85 |
1.51 |
12.94 |
38.219 |
5.289 |
T P |
| 3dyr_A |
111 |
207 |
86 |
1.62 |
13.95 |
38.200 |
5.000 |
T P |
| 1zzy_A |
106 |
207 |
85 |
1.57 |
12.94 |
37.825 |
5.084 |
T P |
| 2gzy_A |
104 |
207 |
83 |
1.52 |
15.66 |
37.721 |
5.116 |
T P |
| 1tho_A |
109 |
207 |
85 |
1.61 |
12.94 |
37.679 |
4.958 |
T P |
| 2tir_A |
108 |
207 |
85 |
1.59 |
12.94 |
37.662 |
5.031 |
T P |
| 2o8v_B |
108 |
207 |
85 |
1.59 |
12.94 |
37.603 |
5.040 |
T P |
| 1quw_A |
105 |
207 |
85 |
1.64 |
14.12 |
37.571 |
4.892 |
T P |
| 2fch_D |
107 |
207 |
86 |
1.72 |
13.95 |
37.560 |
4.714 |
T P |
| 1rqm_A |
105 |
207 |
85 |
1.65 |
14.12 |
37.546 |
4.851 |
T P |
| 2h76_A |
108 |
207 |
85 |
1.61 |
12.94 |
37.452 |
4.977 |
T P |
| 2h6z_A |
108 |
207 |
85 |
1.61 |
12.94 |
37.448 |
4.958 |
T P |
| 2eio_A |
107 |
207 |
84 |
1.56 |
13.10 |
37.444 |
5.062 |
T P |
| 2h74_A |
108 |
207 |
85 |
1.65 |
12.94 |
37.427 |
4.857 |
T P |
| 2cvk_A |
105 |
207 |
85 |
1.72 |
17.65 |
37.399 |
4.667 |
T P |
| 2h6y_A |
107 |
207 |
85 |
1.66 |
12.94 |
37.315 |
4.835 |
T P |
| 2eir_A |
107 |
207 |
84 |
1.56 |
13.10 |
37.306 |
5.065 |
T P |
| 2fd3_A |
108 |
207 |
85 |
1.63 |
12.94 |
37.276 |
4.925 |
T P |
| 2o7k_A |
103 |
207 |
84 |
1.61 |
17.86 |
37.272 |
4.902 |
T P |
| 2h70_A |
107 |
207 |
85 |
1.71 |
12.94 |
37.228 |
4.693 |
T P |
| 1nw2_A |
105 |
207 |
85 |
1.82 |
15.29 |
37.192 |
4.425 |
T P |
| 2h73_A |
108 |
207 |
85 |
1.70 |
14.12 |
37.183 |
4.730 |
T P |
| 1t00_A |
112 |
207 |
82 |
1.57 |
15.85 |
37.171 |
4.912 |
T P |
| 2o87_A |
103 |
207 |
84 |
1.67 |
19.05 |
37.168 |
4.748 |
T P |
| 2h71_A |
107 |
207 |
85 |
1.68 |
12.94 |
37.154 |
4.772 |
T P |
| 2o85_A |
103 |
207 |
84 |
1.69 |
19.05 |
37.135 |
4.706 |
T P |
| 1v98_A |
92 |
207 |
86 |
1.79 |
22.09 |
37.118 |
4.538 |
T P |
| 2eiq_A |
107 |
207 |
84 |
1.62 |
13.10 |
37.111 |
4.883 |
T P |
| 2o89_A |
103 |
207 |
84 |
1.70 |
19.05 |
37.046 |
4.676 |
T P |
| 1keb_A |
108 |
207 |
85 |
1.73 |
12.94 |
37.039 |
4.646 |
T P |
| 2yzu_A |
104 |
207 |
84 |
1.70 |
16.67 |
37.003 |
4.676 |
T P |
| 2h72_A |
107 |
207 |
85 |
1.72 |
12.94 |
36.984 |
4.681 |
T P |
| 1t3b_A |
209 |
207 |
106 |
2.03 |
14.15 |
36.974 |
4.972 |
T P |
| 1nsw_A |
105 |
207 |
85 |
1.82 |
15.29 |
36.707 |
4.422 |
T P |
| 2h75_B |
108 |
207 |
84 |
1.73 |
14.29 |
36.552 |
4.585 |
T P |
| 1f6m_C |
108 |
207 |
85 |
1.78 |
14.12 |
36.543 |
4.511 |
T P |
| 2j23_A |
115 |
207 |
84 |
1.83 |
14.29 |
36.278 |
4.343 |
T P |
| 1v58_A |
229 |
207 |
106 |
2.27 |
16.04 |
36.109 |
4.476 |
T P |
| 1z6m_A |
175 |
207 |
116 |
2.45 |
9.48 |
35.900 |
4.555 |
T P |
| 2i1u_A |
108 |
207 |
82 |
1.80 |
13.41 |
35.762 |
4.321 |
T P |
| 2h0i_A |
229 |
207 |
107 |
2.27 |
16.82 |
35.467 |
4.513 |
T P |
| 2h0g_A |
229 |
207 |
106 |
2.24 |
16.98 |
35.458 |
4.524 |
T P |
| 1x5d_A |
133 |
207 |
83 |
1.99 |
16.87 |
34.833 |
3.974 |
T P |
| 2dj2_A |
120 |
207 |
80 |
1.86 |
16.25 |
34.784 |
4.077 |
T P |
| 3f8u_A |
469 |
207 |
81 |
1.98 |
11.11 |
34.409 |
3.895 |
T P |
| 2h0h_B |
230 |
207 |
104 |
2.36 |
18.27 |
34.373 |
4.233 |
T P |
| 2dml_A |
130 |
207 |
81 |
2.03 |
16.05 |
32.558 |
3.803 |
T P |
| 1f9m_A |
112 |
207 |
83 |
1.84 |
14.46 |
32.538 |
4.286 |
T P |
| 1faa_A |
121 |
207 |
83 |
1.86 |
14.46 |
32.485 |
4.240 |
T P |
| 2pu9_C |
111 |
207 |
83 |
1.90 |
13.25 |
32.282 |
4.155 |
T P |
| 1kng_A |
144 |
207 |
92 |
2.15 |
10.87 |
32.240 |
4.095 |
T P |
| 1z5y_E |
136 |
207 |
87 |
1.97 |
17.24 |
31.586 |
4.203 |
T P |
| 2alb_A |
113 |
207 |
76 |
2.16 |
13.16 |
31.263 |
3.364 |
T P |
| 1srx_A |
108 |
207 |
83 |
2.15 |
12.05 |
30.086 |
3.697 |
T P |
| 1dyv_A |
186 |
207 |
71 |
1.89 |
9.86 |
29.010 |
3.574 |
T P |
| 1mek_A |
120 |
207 |
79 |
2.27 |
10.13 |
25.836 |
3.331 |
T P |
| 1fo5_A |
85 |
207 |
62 |
2.53 |
14.52 |
19.587 |
2.356 |
T P |
| 1nho_A |
85 |
207 |
52 |
2.50 |
15.38 |
16.432 |
2.002 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]