LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_272.5wLII_11233_46
Total number of 3D structures: 39
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
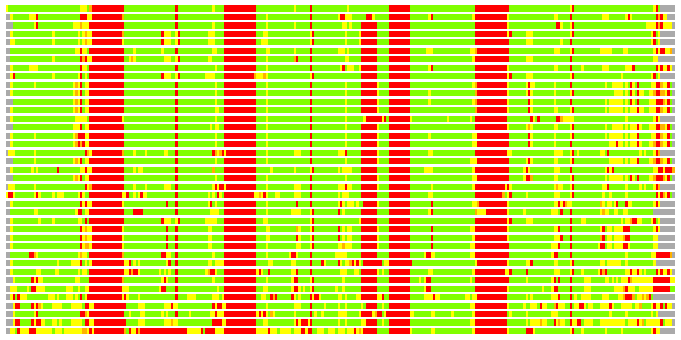
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1erj_C |
357 |
288 |
226 |
0.77 |
17.70 |
77.365 |
26.041 |
T P |
| 3dm0_A |
675 |
288 |
217 |
1.37 |
15.67 |
72.650 |
14.728 |
T P |
| 1vyh_C |
310 |
288 |
217 |
1.33 |
15.21 |
72.644 |
15.227 |
T P |
| 1got_B |
339 |
288 |
217 |
1.36 |
11.98 |
71.858 |
14.832 |
T P |
| 2bcj_B |
339 |
288 |
217 |
1.37 |
12.44 |
71.575 |
14.769 |
T P |
| 3fm0_A |
328 |
288 |
219 |
1.51 |
12.79 |
71.552 |
13.587 |
T P |
| 2pbi_D |
354 |
288 |
214 |
1.25 |
10.75 |
71.540 |
15.880 |
T P |
| 3frx_B |
313 |
288 |
216 |
1.35 |
10.65 |
71.304 |
14.907 |
T P |
| 1tbg_A |
340 |
288 |
214 |
1.40 |
11.68 |
71.012 |
14.242 |
T P |
| 2gnq_A |
316 |
288 |
217 |
1.46 |
12.90 |
70.925 |
13.917 |
T P |
| 2g9a_A |
310 |
288 |
217 |
1.50 |
12.44 |
70.841 |
13.604 |
T P |
| 2h9l_A |
321 |
288 |
215 |
1.42 |
13.02 |
70.830 |
14.181 |
T P |
| 3emh_A |
300 |
288 |
218 |
1.52 |
12.84 |
70.672 |
13.475 |
T P |
| 2g99_A |
304 |
288 |
214 |
1.36 |
12.62 |
70.648 |
14.619 |
T P |
| 2h14_A |
303 |
288 |
217 |
1.50 |
13.36 |
70.602 |
13.570 |
T P |
| 2h9m_A |
304 |
288 |
215 |
1.45 |
12.56 |
70.544 |
13.838 |
T P |
| 2co0_A |
304 |
288 |
215 |
1.47 |
13.49 |
70.498 |
13.691 |
T P |
| 3cfs_B |
383 |
288 |
216 |
1.53 |
9.26 |
70.469 |
13.266 |
T P |
| 2h6n_B |
305 |
288 |
216 |
1.46 |
12.96 |
70.469 |
13.813 |
T P |
| 1gxr_A |
335 |
288 |
214 |
1.45 |
16.36 |
70.189 |
13.837 |
T P |
| 2cnx_A |
306 |
288 |
213 |
1.42 |
12.68 |
70.121 |
13.982 |
T P |
| 3c99_A |
380 |
288 |
214 |
1.51 |
10.28 |
70.108 |
13.262 |
T P |
| 3cfv_B |
393 |
288 |
217 |
1.67 |
8.29 |
69.585 |
12.281 |
T P |
| 3bg1_A |
285 |
288 |
212 |
1.57 |
8.96 |
69.405 |
12.685 |
T P |
| 2hes_X |
308 |
288 |
211 |
1.46 |
11.37 |
69.371 |
13.490 |
T P |
| 1a0r_B |
339 |
288 |
212 |
1.47 |
12.74 |
69.294 |
13.463 |
T P |
| 2pm7_D |
288 |
288 |
210 |
1.47 |
8.10 |
69.294 |
13.349 |
T P |
| 2pm6_D |
288 |
288 |
210 |
1.49 |
8.10 |
69.262 |
13.196 |
T P |
| 2pm9_B |
280 |
288 |
210 |
1.53 |
8.57 |
69.026 |
12.894 |
T P |
| 1p22_A |
402 |
288 |
207 |
1.43 |
11.11 |
68.394 |
13.515 |
T P |
| 1nr0_A |
610 |
288 |
208 |
1.69 |
12.02 |
67.078 |
11.652 |
T P |
| 3ei4_B |
368 |
288 |
207 |
1.77 |
11.11 |
66.300 |
11.051 |
T P |
| 1nex_B |
444 |
288 |
206 |
1.70 |
17.48 |
65.959 |
11.441 |
T P |
| 2ovr_B |
442 |
288 |
207 |
1.70 |
11.59 |
65.489 |
11.529 |
T P |
| 1r5m_A |
351 |
288 |
200 |
1.99 |
13.00 |
60.275 |
9.588 |
T P |
| 2aq5_A |
395 |
288 |
209 |
2.03 |
11.48 |
59.903 |
9.823 |
T P |
| 2pm9_A |
384 |
288 |
209 |
1.75 |
10.05 |
59.340 |
11.298 |
T P |
| 2b4e_A |
386 |
288 |
207 |
2.08 |
10.63 |
54.644 |
9.478 |
T P |
| 2zkq_a |
306 |
288 |
181 |
2.28 |
9.39 |
50.855 |
7.607 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]