LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_280.5wLII_11238_7
Total number of 3D structures: 88
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
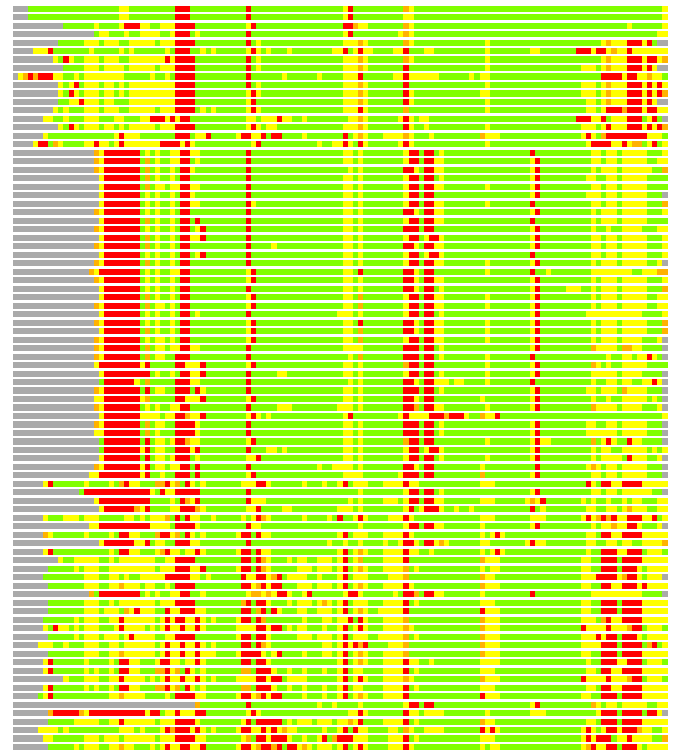
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2b1k_A |
149 |
129 |
121 |
0.77 |
20.66 |
92.551 |
13.884 |
T P |
| 2g0f_A |
149 |
129 |
121 |
0.80 |
19.83 |
92.472 |
13.379 |
T P |
| 1kng_A |
144 |
129 |
109 |
1.48 |
20.18 |
80.420 |
6.899 |
T P |
| 1z5y_E |
136 |
129 |
108 |
1.28 |
20.37 |
79.971 |
7.848 |
T P |
| 1st9_A |
137 |
129 |
109 |
1.84 |
17.43 |
76.462 |
5.612 |
T P |
| 2ywi_A |
190 |
129 |
111 |
1.89 |
14.41 |
76.041 |
5.586 |
T P |
| 2f9s_A |
138 |
129 |
109 |
1.92 |
15.60 |
75.894 |
5.405 |
T P |
| 2h19_A |
138 |
129 |
107 |
1.91 |
15.89 |
74.515 |
5.331 |
T P |
| 1jfu_B |
177 |
129 |
111 |
2.08 |
18.92 |
74.207 |
5.085 |
T P |
| 3c73_B |
138 |
129 |
108 |
1.96 |
15.74 |
74.176 |
5.255 |
T P |
| 2h1a_B |
139 |
129 |
108 |
1.99 |
15.74 |
74.137 |
5.171 |
T P |
| 2h1g_B |
137 |
129 |
107 |
1.92 |
14.95 |
73.958 |
5.293 |
T P |
| 2h1b_A |
138 |
129 |
107 |
1.98 |
15.89 |
73.275 |
5.145 |
T P |
| 3fkf_A |
141 |
129 |
106 |
1.94 |
16.98 |
72.791 |
5.187 |
T P |
| 3c71_A |
140 |
129 |
107 |
2.02 |
15.89 |
72.332 |
5.039 |
T P |
| 1we0_A |
166 |
129 |
103 |
1.85 |
12.62 |
71.785 |
5.285 |
T P |
| 2hyx_A |
333 |
129 |
106 |
1.87 |
20.75 |
70.358 |
5.378 |
T P |
| 2trx_A |
108 |
129 |
98 |
1.63 |
20.41 |
69.995 |
5.677 |
T P |
| 3dyr_A |
111 |
129 |
98 |
1.66 |
20.41 |
69.968 |
5.581 |
T P |
| 2fd3_A |
108 |
129 |
98 |
1.71 |
19.39 |
69.716 |
5.400 |
T P |
| 2eio_A |
107 |
129 |
97 |
1.58 |
20.62 |
69.682 |
5.776 |
T P |
| 3dxb_D |
216 |
129 |
97 |
1.61 |
20.62 |
69.417 |
5.658 |
T P |
| 2eir_A |
107 |
129 |
96 |
1.56 |
20.83 |
69.203 |
5.772 |
T P |
| 2tir_A |
108 |
129 |
97 |
1.68 |
20.62 |
69.189 |
5.459 |
T P |
| 2h73_A |
108 |
129 |
98 |
1.73 |
20.41 |
69.183 |
5.369 |
T P |
| 1txx_A |
108 |
129 |
96 |
1.63 |
19.79 |
69.183 |
5.561 |
T P |
| 2o8v_B |
108 |
129 |
96 |
1.61 |
19.79 |
69.169 |
5.601 |
T P |
| 2fch_D |
107 |
129 |
96 |
1.67 |
19.79 |
69.026 |
5.423 |
T P |
| 1tho_A |
109 |
129 |
98 |
1.73 |
19.39 |
68.979 |
5.360 |
T P |
| 1zzy_A |
106 |
129 |
96 |
1.67 |
20.83 |
68.965 |
5.439 |
T P |
| 2h72_A |
107 |
129 |
97 |
1.72 |
20.62 |
68.934 |
5.332 |
T P |
| 1t00_A |
112 |
129 |
97 |
1.75 |
19.59 |
68.921 |
5.248 |
T P |
| 2h74_A |
108 |
129 |
98 |
1.76 |
20.41 |
68.917 |
5.259 |
T P |
| 2h75_B |
108 |
129 |
97 |
1.69 |
21.65 |
68.904 |
5.422 |
T P |
| 2h76_A |
108 |
129 |
97 |
1.72 |
20.62 |
68.724 |
5.316 |
T P |
| 2h71_A |
107 |
129 |
98 |
1.78 |
19.39 |
68.625 |
5.221 |
T P |
| 1keb_A |
108 |
129 |
97 |
1.69 |
20.62 |
68.625 |
5.415 |
T P |
| 2h70_A |
107 |
129 |
97 |
1.75 |
20.62 |
68.567 |
5.239 |
T P |
| 2h6y_A |
107 |
129 |
97 |
1.76 |
20.62 |
68.537 |
5.217 |
T P |
| 2h6z_A |
108 |
129 |
97 |
1.76 |
20.62 |
68.441 |
5.216 |
T P |
| 2i4a_A |
107 |
129 |
96 |
1.77 |
17.71 |
68.231 |
5.126 |
T P |
| 1nw2_A |
105 |
129 |
94 |
1.51 |
17.02 |
68.003 |
5.832 |
T P |
| 2cvk_A |
105 |
129 |
95 |
1.73 |
22.11 |
67.819 |
5.182 |
T P |
| 1zcp_A |
106 |
129 |
93 |
1.58 |
19.35 |
67.625 |
5.527 |
T P |
| 1nsw_A |
105 |
129 |
95 |
1.59 |
17.89 |
67.391 |
5.631 |
T P |
| 2o7k_A |
103 |
129 |
93 |
1.68 |
18.28 |
67.200 |
5.237 |
T P |
| 2yzu_A |
104 |
129 |
96 |
1.81 |
22.92 |
67.180 |
5.022 |
T P |
| 1f6m_C |
108 |
129 |
97 |
1.88 |
19.59 |
67.166 |
4.887 |
T P |
| 2b1l_B |
129 |
129 |
92 |
1.71 |
19.57 |
67.037 |
5.077 |
T P |
| 2o87_A |
103 |
129 |
94 |
1.74 |
17.02 |
66.891 |
5.098 |
T P |
| 2o85_A |
103 |
129 |
94 |
1.76 |
17.02 |
66.864 |
5.048 |
T P |
| 1xwb_A |
106 |
129 |
93 |
1.74 |
13.98 |
66.816 |
5.041 |
T P |
| 2voc_A |
110 |
129 |
94 |
1.75 |
14.89 |
66.677 |
5.077 |
T P |
| 1xwa_A |
111 |
129 |
93 |
1.77 |
13.98 |
66.541 |
4.963 |
T P |
| 1dby_A |
107 |
129 |
95 |
1.88 |
15.79 |
66.525 |
4.787 |
T P |
| 2gzy_A |
104 |
129 |
93 |
1.73 |
18.28 |
66.306 |
5.074 |
T P |
| 1x0r_A |
244 |
129 |
108 |
2.07 |
13.89 |
66.248 |
4.988 |
T P |
| 2eiq_A |
107 |
129 |
90 |
1.53 |
22.22 |
65.626 |
5.511 |
T P |
| 1srx_A |
108 |
129 |
95 |
1.93 |
20.00 |
65.491 |
4.682 |
T P |
| 1quw_A |
105 |
129 |
96 |
1.97 |
15.62 |
65.342 |
4.646 |
T P |
| 2cx4_A |
161 |
129 |
110 |
2.16 |
17.27 |
65.300 |
4.860 |
T P |
| 2ipa_A |
104 |
129 |
91 |
1.70 |
15.38 |
65.269 |
5.058 |
T P |
| 2zct_D |
238 |
129 |
106 |
1.99 |
12.26 |
64.799 |
5.076 |
T P |
| 1rqm_A |
105 |
129 |
96 |
2.01 |
16.67 |
64.522 |
4.540 |
T P |
| 2e2g_B |
239 |
129 |
106 |
2.01 |
13.21 |
64.258 |
5.020 |
T P |
| 2pn8_A |
191 |
129 |
104 |
2.04 |
11.54 |
63.994 |
4.853 |
T P |
| 2z9s_A |
197 |
129 |
108 |
2.12 |
14.81 |
63.779 |
4.867 |
T P |
| 1zof_B |
175 |
129 |
106 |
2.11 |
15.09 |
63.654 |
4.801 |
T P |
| 2rii_B |
185 |
129 |
105 |
2.11 |
14.29 |
63.528 |
4.760 |
T P |
| 1oaz_A |
115 |
129 |
93 |
2.03 |
19.35 |
63.439 |
4.359 |
T P |
| 1uul_A |
194 |
129 |
106 |
2.01 |
9.43 |
62.995 |
5.023 |
T P |
| 1zye_A |
162 |
129 |
103 |
1.99 |
13.59 |
62.877 |
4.934 |
T P |
| 2i81_A |
193 |
129 |
108 |
2.09 |
14.81 |
62.870 |
4.937 |
T P |
| 2h01_A |
174 |
129 |
108 |
2.18 |
12.04 |
62.869 |
4.743 |
T P |
| 1qmv_A |
196 |
129 |
106 |
2.11 |
14.15 |
62.553 |
4.794 |
T P |
| 1xxu_A |
153 |
129 |
107 |
2.12 |
14.02 |
62.425 |
4.816 |
T P |
| 2c0d_B |
175 |
129 |
106 |
2.04 |
7.55 |
62.284 |
4.959 |
T P |
| 2nvl_A |
242 |
129 |
105 |
1.99 |
12.38 |
62.197 |
5.025 |
T P |
| 2cv4_D |
242 |
129 |
107 |
2.09 |
13.08 |
62.050 |
4.877 |
T P |
| 2feg_A |
169 |
129 |
105 |
2.15 |
12.38 |
61.953 |
4.666 |
T P |
| 1vgs_D |
242 |
129 |
107 |
2.09 |
14.02 |
61.880 |
4.876 |
T P |
| 1yep_A |
168 |
129 |
104 |
1.93 |
13.46 |
61.876 |
5.123 |
T P |
| 1v98_A |
92 |
129 |
87 |
1.66 |
19.54 |
61.842 |
4.940 |
T P |
| 3erw_A |
134 |
129 |
88 |
1.90 |
17.05 |
61.294 |
4.392 |
T P |
| 1e2y_G |
173 |
129 |
102 |
2.05 |
13.73 |
59.913 |
4.751 |
T P |
| 2bmx_B |
178 |
129 |
105 |
2.25 |
15.24 |
58.895 |
4.473 |
T P |
| 2ywn_A |
150 |
129 |
104 |
2.17 |
16.35 |
58.559 |
4.576 |
T P |
| 1qq2_A |
173 |
129 |
102 |
2.20 |
13.73 |
58.491 |
4.426 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]