LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_283.5wLII_11238_16
Total number of 3D structures: 50
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
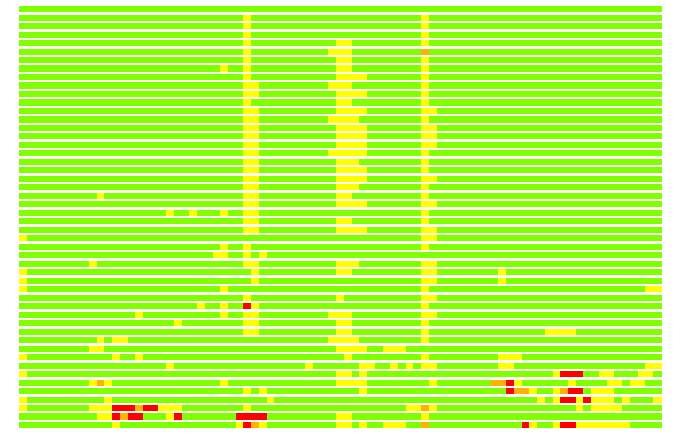
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2gzy_A |
104 |
83 |
83 |
0.27 |
20.48 |
99.905 |
22.161 |
T P |
| 2o87_A |
103 |
83 |
83 |
0.88 |
19.28 |
98.224 |
8.454 |
T P |
| 2o85_A |
103 |
83 |
83 |
0.90 |
18.07 |
98.140 |
8.338 |
T P |
| 2o7k_A |
103 |
83 |
83 |
0.91 |
18.07 |
97.759 |
8.213 |
T P |
| 2trx_A |
108 |
83 |
83 |
1.04 |
14.46 |
97.691 |
7.265 |
T P |
| 2o8v_B |
108 |
83 |
83 |
1.07 |
13.25 |
97.596 |
7.112 |
T P |
| 2i4a_A |
107 |
83 |
83 |
1.05 |
13.25 |
97.432 |
7.189 |
T P |
| 2eio_A |
107 |
83 |
83 |
1.08 |
14.46 |
97.416 |
7.063 |
T P |
| 2fd3_A |
108 |
83 |
83 |
1.10 |
14.46 |
97.331 |
6.927 |
T P |
| 2eir_A |
107 |
83 |
83 |
1.07 |
14.46 |
97.331 |
7.080 |
T P |
| 2h74_A |
108 |
83 |
83 |
1.08 |
14.46 |
97.247 |
7.021 |
T P |
| 2eiq_A |
107 |
83 |
83 |
1.05 |
14.46 |
97.220 |
7.190 |
T P |
| 2h71_A |
107 |
83 |
83 |
1.13 |
14.46 |
97.162 |
6.755 |
T P |
| 3dyr_A |
111 |
83 |
83 |
1.11 |
14.46 |
97.141 |
6.832 |
T P |
| 2h76_A |
108 |
83 |
83 |
1.14 |
14.46 |
97.057 |
6.719 |
T P |
| 2h6y_A |
107 |
83 |
83 |
1.14 |
15.66 |
97.057 |
6.703 |
T P |
| 2h70_A |
107 |
83 |
83 |
1.11 |
14.46 |
97.057 |
6.868 |
T P |
| 2fch_D |
107 |
83 |
83 |
1.14 |
15.66 |
97.046 |
6.699 |
T P |
| 1txx_A |
108 |
83 |
83 |
1.09 |
14.46 |
96.972 |
6.987 |
T P |
| 2h73_A |
108 |
83 |
83 |
1.10 |
14.46 |
96.962 |
6.913 |
T P |
| 2h72_A |
107 |
83 |
83 |
1.13 |
14.46 |
96.888 |
6.726 |
T P |
| 1zzy_A |
106 |
83 |
83 |
1.15 |
14.46 |
96.792 |
6.617 |
T P |
| 1tho_A |
109 |
83 |
83 |
1.09 |
14.46 |
96.713 |
6.951 |
T P |
| 2h6z_A |
108 |
83 |
83 |
1.16 |
14.46 |
96.708 |
6.597 |
T P |
| 1v98_A |
92 |
83 |
83 |
1.08 |
19.28 |
96.655 |
7.037 |
T P |
| 1zcp_A |
106 |
83 |
83 |
1.15 |
13.25 |
96.586 |
6.638 |
T P |
| 3dxb_D |
216 |
83 |
83 |
1.17 |
14.46 |
96.518 |
6.555 |
T P |
| 1nw2_A |
105 |
83 |
83 |
1.12 |
13.25 |
96.375 |
6.824 |
T P |
| 1fb6_A |
104 |
83 |
83 |
1.07 |
14.46 |
96.343 |
7.069 |
T P |
| 1dby_A |
107 |
83 |
83 |
1.12 |
12.05 |
96.285 |
6.818 |
T P |
| 2tir_A |
108 |
83 |
83 |
1.20 |
14.46 |
96.280 |
6.389 |
T P |
| 1nsw_A |
105 |
83 |
83 |
1.14 |
13.25 |
96.185 |
6.692 |
T P |
| 1t00_A |
112 |
83 |
83 |
1.09 |
12.05 |
96.185 |
6.983 |
T P |
| 2i1u_A |
108 |
83 |
83 |
1.15 |
14.46 |
96.074 |
6.623 |
T P |
| 2puk_C |
106 |
83 |
83 |
1.16 |
13.25 |
95.899 |
6.575 |
T P |
| 1m7t_A |
107 |
83 |
82 |
1.07 |
20.73 |
95.619 |
7.016 |
T P |
| 1f6m_C |
108 |
83 |
83 |
1.26 |
13.25 |
95.408 |
6.104 |
T P |
| 1keb_A |
108 |
83 |
83 |
1.27 |
15.66 |
95.313 |
6.074 |
T P |
| 2h75_B |
108 |
83 |
83 |
1.32 |
14.46 |
95.265 |
5.864 |
T P |
| 2voc_A |
110 |
83 |
83 |
1.32 |
19.28 |
94.858 |
5.861 |
T P |
| 2ipa_A |
104 |
83 |
83 |
1.40 |
19.28 |
94.383 |
5.519 |
T P |
| 1quw_A |
105 |
83 |
83 |
1.33 |
13.25 |
94.193 |
5.784 |
T P |
| 1rqm_A |
105 |
83 |
83 |
1.41 |
13.25 |
92.903 |
5.510 |
T P |
| 3f8u_A |
469 |
83 |
80 |
1.46 |
10.00 |
90.694 |
5.141 |
T P |
| 1srx_A |
108 |
83 |
82 |
1.72 |
13.41 |
89.717 |
4.502 |
T P |
| 1x5d_A |
133 |
83 |
80 |
1.66 |
17.50 |
89.453 |
4.535 |
T P |
| 2alb_A |
113 |
83 |
80 |
1.57 |
10.00 |
88.153 |
4.779 |
T P |
| 1gl8_A |
104 |
83 |
78 |
1.80 |
14.10 |
85.484 |
4.104 |
T P |
| 1oaz_A |
115 |
83 |
75 |
1.44 |
16.00 |
85.082 |
4.870 |
T P |
| 1mek_A |
120 |
83 |
79 |
1.79 |
16.46 |
82.399 |
4.177 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]