LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_284.5wLII_11238_18
Total number of 3D structures: 52
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
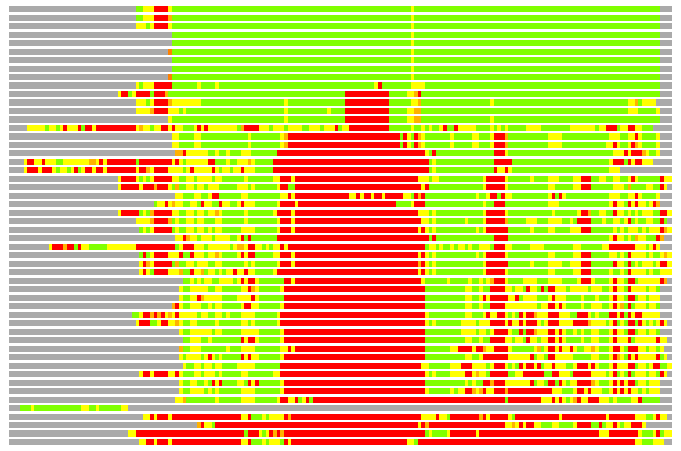
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2qjp_C |
179 |
183 |
141 |
0.76 |
19.15 |
76.397 |
16.433 |
T P |
| 2qjk_C |
179 |
183 |
141 |
0.77 |
19.86 |
76.309 |
16.179 |
T P |
| 2qjy_C |
179 |
183 |
141 |
0.75 |
19.86 |
76.158 |
16.668 |
T P |
| 2nve_A |
141 |
183 |
135 |
0.52 |
18.52 |
73.895 |
21.842 |
T P |
| 2nuk_A |
141 |
183 |
135 |
0.51 |
19.26 |
73.895 |
22.054 |
T P |
| 2num_A |
141 |
183 |
136 |
0.75 |
18.38 |
73.886 |
16.008 |
T P |
| 2nvf_A |
141 |
183 |
135 |
0.51 |
18.52 |
73.857 |
22.097 |
T P |
| 2nvg_A |
141 |
183 |
135 |
0.50 |
18.52 |
73.857 |
22.316 |
T P |
| 2nwf_A |
141 |
183 |
136 |
0.76 |
18.38 |
73.842 |
15.892 |
T P |
| 1zrt_E |
181 |
183 |
139 |
1.07 |
20.14 |
73.708 |
11.833 |
T P |
| 1bcc_E |
196 |
183 |
128 |
1.21 |
20.31 |
67.692 |
9.780 |
T P |
| 1pp9_E |
196 |
183 |
128 |
1.54 |
21.09 |
67.117 |
7.795 |
T P |
| 3cwb_E |
196 |
183 |
129 |
1.50 |
20.93 |
65.756 |
8.065 |
T P |
| 1rie_A |
127 |
183 |
124 |
1.14 |
20.16 |
65.653 |
10.009 |
T P |
| 3cx5_E |
185 |
183 |
130 |
2.46 |
20.00 |
56.983 |
5.074 |
T P |
| 1g8k_B |
133 |
183 |
98 |
1.83 |
23.47 |
47.420 |
5.072 |
T P |
| 1g8j_D |
129 |
183 |
97 |
1.89 |
23.71 |
46.372 |
4.886 |
T P |
| 1rfs_A |
127 |
183 |
84 |
1.78 |
22.62 |
42.671 |
4.473 |
T P |
| 2e74_D |
166 |
183 |
96 |
2.21 |
26.04 |
42.259 |
4.163 |
T P |
| 2zt9_D |
166 |
183 |
92 |
2.21 |
22.83 |
41.032 |
3.986 |
T P |
| 1uli_C |
425 |
183 |
97 |
2.10 |
19.59 |
40.528 |
4.413 |
T P |
| 2b1x_A |
441 |
183 |
94 |
1.96 |
14.89 |
40.388 |
4.564 |
T P |
| 1jm1_A |
202 |
183 |
98 |
1.96 |
23.47 |
40.246 |
4.769 |
T P |
| 1nyk_A |
156 |
183 |
95 |
1.97 |
27.37 |
39.586 |
4.585 |
T P |
| 1wql_A |
436 |
183 |
95 |
2.13 |
15.79 |
39.042 |
4.259 |
T P |
| 2gbw_A |
449 |
183 |
94 |
2.19 |
15.96 |
38.511 |
4.111 |
T P |
| 2bmo_A |
437 |
183 |
94 |
2.15 |
18.09 |
38.027 |
4.185 |
T P |
| 1q90_C |
126 |
183 |
83 |
1.84 |
25.30 |
37.905 |
4.281 |
T P |
| 1um3_D |
168 |
183 |
97 |
2.38 |
22.68 |
37.361 |
3.911 |
T P |
| 2hmj_A |
446 |
183 |
90 |
2.10 |
14.44 |
37.191 |
4.100 |
T P |
| 2ckf_A |
448 |
183 |
90 |
2.12 |
14.44 |
36.655 |
4.049 |
T P |
| 1o7n_A |
447 |
183 |
90 |
2.16 |
14.44 |
35.513 |
3.987 |
T P |
| 1vm9_A |
109 |
183 |
84 |
2.21 |
11.90 |
34.946 |
3.637 |
T P |
| 3dqy_A |
106 |
183 |
83 |
2.22 |
14.46 |
33.737 |
3.580 |
T P |
| 1fqt_A |
109 |
183 |
81 |
2.23 |
17.28 |
33.444 |
3.477 |
T P |
| 2de6_E |
106 |
183 |
84 |
2.29 |
14.29 |
33.268 |
3.510 |
T P |
| 2de6_B |
390 |
183 |
83 |
2.49 |
14.46 |
32.262 |
3.205 |
T P |
| 1z01_A |
427 |
183 |
81 |
2.36 |
9.88 |
31.492 |
3.290 |
T P |
| 2e4q_A |
108 |
183 |
81 |
2.26 |
12.35 |
31.045 |
3.432 |
T P |
| 2qpz_A |
103 |
183 |
81 |
2.23 |
8.64 |
30.944 |
3.479 |
T P |
| 1sjg_A |
112 |
183 |
78 |
2.32 |
7.69 |
30.037 |
3.218 |
T P |
| 2i7f_A |
102 |
183 |
79 |
2.35 |
18.99 |
29.771 |
3.228 |
T P |
| 3c0d_A |
109 |
183 |
76 |
2.28 |
6.58 |
28.652 |
3.191 |
T P |
| 2zyl_A |
359 |
183 |
76 |
2.39 |
10.53 |
28.048 |
3.056 |
T P |
| 3d89_A |
136 |
183 |
73 |
2.36 |
12.33 |
27.620 |
2.969 |
T P |
| 2jo6_A |
110 |
183 |
70 |
2.35 |
12.86 |
26.291 |
2.861 |
T P |
| 2jza_A |
130 |
183 |
61 |
2.16 |
8.20 |
24.260 |
2.702 |
T P |
| 1q90_R |
39 |
183 |
30 |
1.57 |
20.00 |
15.595 |
1.800 |
T P |
| 1dkx_A |
219 |
183 |
37 |
2.76 |
10.81 |
12.211 |
1.294 |
T P |
| 2op6_A |
150 |
183 |
33 |
2.71 |
12.12 |
12.126 |
1.174 |
T P |
| 3dpo_B |
215 |
183 |
27 |
2.50 |
11.11 |
10.378 |
1.039 |
T P |
| 1dkz_A |
215 |
183 |
27 |
2.67 |
11.11 |
9.575 |
0.974 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]