LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_285.5wLII_11238_20
Total number of 3D structures: 61
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
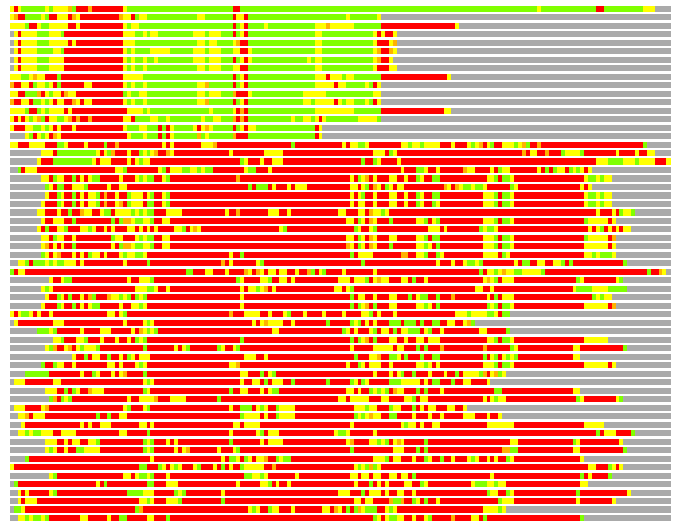
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1pj5_A |
827 |
169 |
149 |
1.23 |
14.77 |
86.344 |
11.213 |
T P |
| 1y56_B |
374 |
169 |
77 |
1.87 |
7.79 |
40.085 |
3.907 |
T P |
| 1x31_B |
402 |
169 |
79 |
2.03 |
11.39 |
38.552 |
3.715 |
T P |
| 1b3m_A |
385 |
169 |
77 |
1.90 |
6.49 |
38.348 |
3.858 |
T P |
| 3bhk_A |
385 |
169 |
79 |
2.01 |
8.86 |
38.234 |
3.753 |
T P |
| 1l9c_A |
385 |
169 |
78 |
2.02 |
7.69 |
37.948 |
3.688 |
T P |
| 2gf3_A |
385 |
169 |
77 |
1.95 |
7.79 |
37.890 |
3.761 |
T P |
| 1el9_A |
385 |
169 |
77 |
1.96 |
7.79 |
37.886 |
3.730 |
T P |
| 2gah_B |
403 |
169 |
74 |
2.03 |
10.81 |
37.399 |
3.472 |
T P |
| 1ng4_A |
364 |
169 |
73 |
2.00 |
10.96 |
36.946 |
3.477 |
T P |
| 1zov_A |
381 |
169 |
76 |
2.12 |
6.58 |
36.883 |
3.423 |
T P |
| 1ryi_A |
364 |
169 |
76 |
2.14 |
14.47 |
36.612 |
3.394 |
T P |
| 2gag_B |
403 |
169 |
78 |
2.22 |
11.54 |
35.744 |
3.364 |
T P |
| 2uzz_B |
372 |
169 |
77 |
2.28 |
6.49 |
33.085 |
3.229 |
T P |
| 1aa8_A |
340 |
169 |
55 |
2.01 |
9.09 |
26.803 |
2.611 |
T P |
| 1an9_A |
340 |
169 |
51 |
1.89 |
13.73 |
26.454 |
2.558 |
T P |
| 3dhy_A |
486 |
169 |
51 |
2.80 |
7.84 |
19.962 |
1.761 |
T P |
| 3g79_A |
475 |
169 |
50 |
2.67 |
4.00 |
19.385 |
1.805 |
T P |
| 2q3e_A |
460 |
169 |
46 |
2.43 |
2.17 |
19.365 |
1.820 |
T P |
| 3ce6_A |
486 |
169 |
46 |
2.60 |
8.70 |
18.380 |
1.702 |
T P |
| 2iat_A |
312 |
169 |
44 |
2.62 |
4.55 |
17.579 |
1.620 |
T P |
| 1n7d_A |
639 |
169 |
44 |
2.58 |
6.82 |
17.364 |
1.641 |
T P |
| 2iao_A |
312 |
169 |
43 |
2.62 |
4.65 |
17.324 |
1.581 |
T P |
| 2iax_A |
312 |
169 |
44 |
2.72 |
4.55 |
17.090 |
1.559 |
T P |
| 2ecf_A |
700 |
169 |
45 |
2.68 |
6.67 |
17.034 |
1.619 |
T P |
| 2iaq_A |
312 |
169 |
43 |
2.77 |
11.63 |
16.795 |
1.499 |
T P |
| 1dli_A |
402 |
169 |
44 |
2.78 |
4.55 |
16.786 |
1.525 |
T P |
| 2iar_A |
312 |
169 |
43 |
2.76 |
9.30 |
16.722 |
1.503 |
T P |
| 2ias_A |
312 |
169 |
43 |
2.85 |
11.63 |
16.667 |
1.458 |
T P |
| 2iaw_A |
312 |
169 |
45 |
2.77 |
6.67 |
16.313 |
1.570 |
T P |
| 1mv8_A |
436 |
169 |
39 |
2.50 |
7.69 |
16.127 |
1.500 |
T P |
| 2o3j_A |
465 |
169 |
42 |
2.87 |
9.52 |
15.819 |
1.412 |
T P |
| 2h6n_B |
305 |
169 |
43 |
2.79 |
4.65 |
15.680 |
1.490 |
T P |
| 3dr2_A |
299 |
169 |
38 |
2.63 |
5.26 |
15.545 |
1.394 |
T P |
| 2gvu_A |
312 |
169 |
39 |
2.61 |
2.56 |
15.528 |
1.437 |
T P |
| 2iav_A |
312 |
169 |
39 |
2.67 |
10.26 |
15.496 |
1.409 |
T P |
| 1q7f_B |
282 |
169 |
40 |
2.70 |
5.00 |
15.247 |
1.430 |
T P |
| 1pjx_A |
314 |
169 |
36 |
2.54 |
8.33 |
15.137 |
1.362 |
T P |
| 2dso_C |
323 |
169 |
36 |
2.61 |
5.56 |
15.101 |
1.327 |
T P |
| 2gvx_A |
312 |
169 |
38 |
2.79 |
7.89 |
14.892 |
1.315 |
T P |
| 2h14_A |
303 |
169 |
36 |
2.61 |
8.33 |
14.817 |
1.326 |
T P |
| 2dg0_A |
322 |
169 |
35 |
2.58 |
8.57 |
14.779 |
1.304 |
T P |
| 2iap_A |
312 |
169 |
40 |
2.96 |
7.50 |
14.771 |
1.306 |
T P |
| 2dg1_B |
322 |
169 |
36 |
2.59 |
5.56 |
14.649 |
1.340 |
T P |
| 2iau_A |
312 |
169 |
36 |
2.65 |
8.33 |
14.597 |
1.308 |
T P |
| 3emh_A |
300 |
169 |
37 |
2.75 |
8.11 |
14.488 |
1.298 |
T P |
| 2h9l_A |
321 |
169 |
39 |
2.90 |
10.26 |
14.236 |
1.302 |
T P |
| 2cnx_A |
306 |
169 |
37 |
2.71 |
8.11 |
14.205 |
1.319 |
T P |
| 2g99_A |
304 |
169 |
38 |
2.84 |
5.26 |
14.146 |
1.292 |
T P |
| 3bws_A |
407 |
169 |
35 |
2.90 |
8.57 |
13.600 |
1.167 |
T P |
| 1muu_A |
436 |
169 |
32 |
2.64 |
18.75 |
13.596 |
1.169 |
T P |
| 2co0_A |
304 |
169 |
36 |
2.94 |
5.56 |
13.567 |
1.182 |
T P |
| 2h9m_A |
304 |
169 |
32 |
2.70 |
6.25 |
13.515 |
1.142 |
T P |
| 2ghs_A |
295 |
169 |
34 |
2.77 |
11.76 |
13.507 |
1.183 |
T P |
| 1dlj_A |
402 |
169 |
32 |
2.70 |
9.38 |
13.295 |
1.144 |
T P |
| 3e5z_A |
290 |
169 |
32 |
2.84 |
12.50 |
13.012 |
1.087 |
T P |
| 1l0q_A |
391 |
169 |
33 |
2.66 |
6.06 |
12.891 |
1.194 |
T P |
| 1rwi_A |
256 |
169 |
33 |
2.66 |
6.06 |
12.728 |
1.196 |
T P |
| 1npe_A |
263 |
169 |
31 |
2.84 |
3.23 |
12.644 |
1.053 |
T P |
| 1ijq_A |
308 |
169 |
32 |
2.62 |
3.12 |
12.456 |
1.179 |
T P |
| 2g9a_A |
310 |
169 |
31 |
2.32 |
6.45 |
12.436 |
1.279 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]