LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_286.5wLII_11238_21
Total number of 3D structures: 57
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
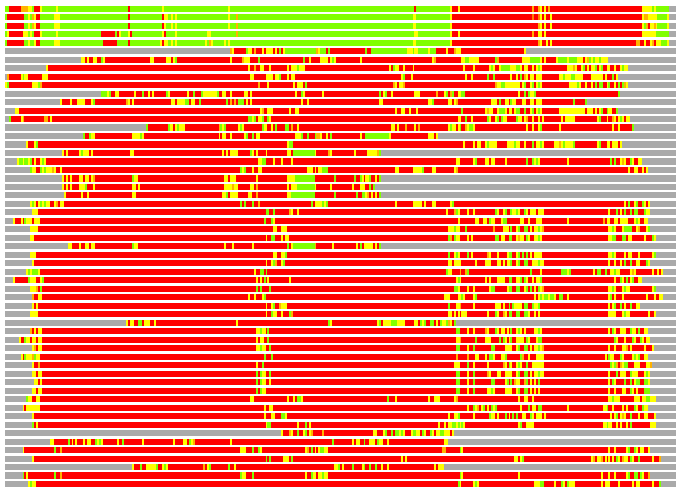
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2nvu_A |
530 |
342 |
237 |
1.34 |
18.57 |
68.266 |
16.411 |
T P |
| 1ngv_A |
529 |
342 |
234 |
1.50 |
18.80 |
65.968 |
14.641 |
T P |
| 1yov_A |
529 |
342 |
232 |
1.37 |
18.97 |
65.619 |
15.774 |
T P |
| 1r4m_A |
516 |
342 |
227 |
1.32 |
18.50 |
64.770 |
16.040 |
T P |
| 1tt5_A |
522 |
342 |
224 |
1.39 |
19.20 |
63.113 |
15.009 |
T P |
| 1y8q_A |
313 |
342 |
76 |
2.18 |
7.89 |
18.450 |
3.338 |
T P |
| 3cmm_C |
1006 |
342 |
80 |
2.64 |
5.00 |
15.316 |
2.925 |
T P |
| 2eyu_A |
247 |
342 |
63 |
2.84 |
6.35 |
11.282 |
2.145 |
T P |
| 1g6o_A |
323 |
342 |
58 |
2.76 |
3.45 |
10.789 |
2.025 |
T P |
| 2eww_A |
343 |
342 |
57 |
2.78 |
5.26 |
10.783 |
1.980 |
T P |
| 1p9r_A |
378 |
342 |
57 |
2.62 |
10.53 |
10.530 |
2.097 |
T P |
| 2it1_A |
362 |
342 |
59 |
2.76 |
5.08 |
10.428 |
2.065 |
T P |
| 2gza_C |
336 |
342 |
59 |
2.95 |
5.08 |
10.283 |
1.936 |
T P |
| 2ewv_A |
343 |
342 |
50 |
2.69 |
6.00 |
9.970 |
1.793 |
T P |
| 2gsz_A |
343 |
342 |
50 |
2.75 |
2.00 |
9.865 |
1.756 |
T P |
| 2d27_A |
148 |
342 |
48 |
2.41 |
10.42 |
9.557 |
1.914 |
T P |
| 2pt7_A |
323 |
342 |
48 |
2.84 |
0.00 |
9.250 |
1.635 |
T P |
| 2hur_A |
142 |
342 |
44 |
2.72 |
6.82 |
8.764 |
1.559 |
T P |
| 1nhk_R |
144 |
342 |
43 |
2.68 |
11.63 |
8.633 |
1.545 |
T P |
| 1f6t_B |
151 |
342 |
44 |
2.87 |
13.64 |
8.445 |
1.480 |
T P |
| 1s57_A |
153 |
342 |
41 |
2.70 |
7.32 |
8.330 |
1.464 |
T P |
| 1xiq_A |
149 |
342 |
42 |
2.72 |
9.52 |
8.282 |
1.487 |
T P |
| 1pae_X |
147 |
342 |
38 |
2.58 |
10.53 |
8.280 |
1.419 |
T P |
| 1nsq_A |
151 |
342 |
43 |
2.70 |
6.98 |
8.249 |
1.535 |
T P |
| 1lwx_A |
150 |
342 |
42 |
2.64 |
11.90 |
8.213 |
1.533 |
T P |
| 2vu5_A |
148 |
342 |
44 |
2.78 |
6.82 |
8.148 |
1.526 |
T P |
| 1leo_A |
150 |
342 |
44 |
2.77 |
9.09 |
8.140 |
1.533 |
T P |
| 3bbb_A |
151 |
342 |
42 |
2.72 |
2.38 |
8.089 |
1.490 |
T P |
| 1k44_A |
135 |
342 |
37 |
2.58 |
16.22 |
7.986 |
1.378 |
T P |
| 1hlw_A |
150 |
342 |
39 |
2.65 |
10.26 |
7.880 |
1.418 |
T P |
| 2hvd_A |
151 |
342 |
42 |
2.92 |
2.38 |
7.856 |
1.389 |
T P |
| 1wkj_A |
137 |
342 |
39 |
2.60 |
2.56 |
7.832 |
1.445 |
T P |
| 1ndl_A |
152 |
342 |
40 |
2.78 |
5.00 |
7.825 |
1.390 |
T P |
| 1w7w_A |
151 |
342 |
38 |
2.55 |
5.26 |
7.814 |
1.434 |
T P |
| 1u8w_A |
149 |
342 |
37 |
2.58 |
2.70 |
7.811 |
1.378 |
T P |
| 1ehw_A |
143 |
342 |
38 |
2.80 |
7.89 |
7.798 |
1.310 |
T P |
| 1bhn_A |
151 |
342 |
42 |
2.78 |
7.14 |
7.797 |
1.457 |
T P |
| 2d28_C |
144 |
342 |
40 |
2.56 |
7.50 |
7.797 |
1.505 |
T P |
| 1npk_A |
150 |
342 |
42 |
2.85 |
14.29 |
7.759 |
1.426 |
T P |
| 1be4_A |
151 |
342 |
42 |
2.77 |
4.76 |
7.753 |
1.463 |
T P |
| 3bbc_A |
151 |
342 |
38 |
2.79 |
10.53 |
7.696 |
1.314 |
T P |
| 2az3_C |
153 |
342 |
41 |
2.79 |
4.88 |
7.654 |
1.420 |
T P |
| 1hhq_A |
150 |
342 |
38 |
2.83 |
5.26 |
7.653 |
1.297 |
T P |
| 1ncl_A |
150 |
342 |
43 |
2.75 |
13.95 |
7.621 |
1.510 |
T P |
| 1nsp_A |
149 |
342 |
36 |
2.54 |
19.44 |
7.585 |
1.364 |
T P |
| 3b54_A |
145 |
342 |
37 |
2.65 |
10.81 |
7.511 |
1.347 |
T P |
| 1ucn_A |
151 |
342 |
40 |
3.01 |
2.50 |
7.510 |
1.286 |
T P |
| 1mn7_A |
150 |
342 |
39 |
2.86 |
10.26 |
7.455 |
1.317 |
T P |
| 2dxe_B |
156 |
342 |
39 |
2.83 |
7.69 |
7.327 |
1.330 |
T P |
| 1ndk_A |
148 |
342 |
37 |
2.72 |
10.81 |
7.076 |
1.311 |
T P |
| 2bh1_X |
68 |
342 |
36 |
2.56 |
8.33 |
6.916 |
1.353 |
T P |
| 1nsk_R |
151 |
342 |
36 |
2.98 |
5.56 |
6.844 |
1.170 |
T P |
| 1zs6_A |
152 |
342 |
31 |
2.84 |
9.68 |
6.270 |
1.053 |
T P |
| 2hve_A |
151 |
342 |
31 |
2.90 |
3.23 |
6.163 |
1.033 |
T P |
| 1pku_A |
149 |
342 |
31 |
2.71 |
6.45 |
6.140 |
1.102 |
T P |
| 2az1_A |
155 |
342 |
30 |
2.65 |
6.67 |
6.008 |
1.090 |
T P |
| 1f3f_A |
150 |
342 |
31 |
2.69 |
9.68 |
5.945 |
1.112 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]