LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_291.5wLII_11238_39
Total number of 3D structures: 38
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
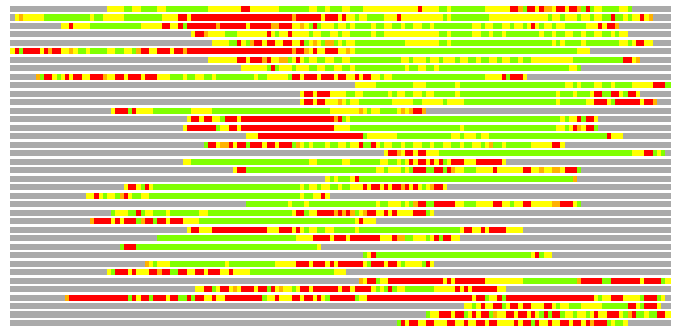
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 2fxm_A |
126 |
157 |
112 |
2.40 |
8.93 |
53.692 |
4.486 |
T P |
| 1c1g_A |
284 |
157 |
113 |
2.33 |
8.85 |
53.349 |
4.648 |
T P |
| 2b9c_B |
142 |
157 |
103 |
2.35 |
5.83 |
51.519 |
4.197 |
T P |
| 2d3e_A |
130 |
157 |
100 |
2.33 |
10.00 |
51.294 |
4.111 |
T P |
| 2v71_A |
160 |
157 |
95 |
2.06 |
6.32 |
51.089 |
4.396 |
T P |
| 2efr_A |
155 |
157 |
92 |
2.19 |
10.87 |
49.623 |
4.026 |
T P |
| 2fxo_A |
129 |
157 |
94 |
2.32 |
11.70 |
48.382 |
3.879 |
T P |
| 1f5n_A |
570 |
157 |
80 |
1.76 |
15.00 |
47.377 |
4.308 |
T P |
| 2zuo_A |
812 |
157 |
84 |
2.23 |
8.33 |
43.222 |
3.607 |
T P |
| 1y1u_A |
544 |
157 |
72 |
1.68 |
6.94 |
42.630 |
4.045 |
T P |
| 2b5u_A |
470 |
157 |
70 |
1.83 |
5.71 |
41.356 |
3.621 |
T P |
| 1jch_A |
468 |
157 |
70 |
2.03 |
5.71 |
40.386 |
3.292 |
T P |
| 1ei3_A |
159 |
157 |
69 |
2.19 |
4.35 |
38.924 |
3.020 |
T P |
| 2hko_A |
647 |
157 |
65 |
1.65 |
10.77 |
38.859 |
3.708 |
T P |
| 2h94_A |
647 |
157 |
65 |
1.81 |
10.77 |
38.790 |
3.412 |
T P |
| 2iw5_A |
666 |
157 |
64 |
1.87 |
9.38 |
37.795 |
3.253 |
T P |
| 1m1j_D |
194 |
157 |
69 |
2.42 |
7.25 |
36.544 |
2.739 |
T P |
| 2ve7_B |
303 |
157 |
59 |
1.56 |
11.86 |
36.095 |
3.562 |
T P |
| 3cwg_B |
507 |
157 |
63 |
1.93 |
6.35 |
36.092 |
3.105 |
T P |
| 2z3y_A |
643 |
157 |
69 |
2.24 |
2.90 |
35.963 |
2.951 |
T P |
| 1qvr_A |
803 |
157 |
59 |
1.36 |
5.08 |
35.923 |
4.050 |
T P |
| 1bg1_A |
559 |
157 |
64 |
1.92 |
6.25 |
35.361 |
3.167 |
T P |
| 1zvu_A |
685 |
157 |
55 |
1.66 |
3.64 |
33.252 |
3.122 |
T P |
| 2dw4_A |
634 |
157 |
66 |
2.18 |
3.03 |
32.100 |
2.896 |
T P |
| 2v1d_A |
666 |
157 |
61 |
2.28 |
4.92 |
31.818 |
2.560 |
T P |
| 3d2f_A |
629 |
157 |
52 |
1.80 |
1.92 |
31.706 |
2.743 |
T P |
| 1ha0_A |
494 |
157 |
54 |
2.03 |
1.85 |
29.450 |
2.539 |
T P |
| 1ses_A |
421 |
157 |
54 |
2.01 |
3.70 |
28.238 |
2.562 |
T P |
| 1h6k_C |
733 |
157 |
45 |
1.12 |
11.11 |
27.581 |
3.697 |
T P |
| 2pms_C |
109 |
157 |
43 |
1.18 |
2.33 |
26.665 |
3.347 |
T P |
| 1wle_B |
469 |
157 |
50 |
2.20 |
8.00 |
25.990 |
2.172 |
T P |
| 1lrz_A |
400 |
157 |
41 |
2.20 |
7.32 |
21.127 |
1.779 |
T P |
| 2v5d_A |
722 |
157 |
35 |
2.41 |
2.86 |
17.826 |
1.395 |
T P |
| 1gax_A |
862 |
157 |
41 |
2.69 |
7.32 |
17.452 |
1.468 |
T P |
| 2qzv_A |
749 |
157 |
41 |
2.82 |
7.32 |
16.756 |
1.403 |
T P |
| 1giq_A |
411 |
157 |
33 |
2.61 |
3.03 |
15.428 |
1.220 |
T P |
| 2j62_A |
585 |
157 |
35 |
2.66 |
5.71 |
14.427 |
1.269 |
T P |
| 2v5c_A |
584 |
157 |
30 |
2.81 |
3.33 |
12.937 |
1.032 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]