LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_293.5wLII_11238_43
Total number of 3D structures: 28
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
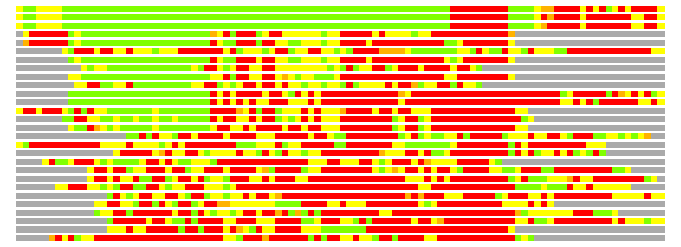
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1c30_A |
1058 |
100 |
80 |
1.76 |
16.25 |
75.518 |
4.303 |
T P |
| 1a9x_A |
1058 |
100 |
77 |
1.66 |
15.58 |
73.197 |
4.368 |
T P |
| 1m6v_A |
1059 |
100 |
77 |
1.59 |
15.58 |
72.785 |
4.556 |
T P |
| 2z3y_A |
643 |
100 |
46 |
2.21 |
8.70 |
36.682 |
1.993 |
T P |
| 2iw5_A |
666 |
100 |
42 |
2.03 |
9.52 |
36.351 |
1.976 |
T P |
| 3cgh_A |
507 |
100 |
55 |
2.70 |
1.82 |
36.062 |
1.963 |
T P |
| 2v1d_A |
666 |
100 |
41 |
2.05 |
9.76 |
32.798 |
1.905 |
T P |
| 1x13_A |
368 |
100 |
44 |
2.32 |
2.27 |
31.653 |
1.816 |
T P |
| 2h94_A |
647 |
100 |
40 |
2.09 |
7.50 |
31.382 |
1.826 |
T P |
| 1ptj_B |
366 |
100 |
46 |
2.38 |
10.87 |
31.037 |
1.857 |
T P |
| 2hko_A |
647 |
100 |
39 |
2.16 |
12.82 |
30.887 |
1.725 |
T P |
| 2dw4_A |
634 |
100 |
39 |
2.18 |
20.51 |
29.581 |
1.707 |
T P |
| 2frd_B |
362 |
100 |
42 |
2.47 |
2.38 |
29.405 |
1.637 |
T P |
| 2fsv_A |
366 |
100 |
39 |
2.26 |
2.56 |
29.063 |
1.655 |
T P |
| 1nm5_A |
360 |
100 |
36 |
2.31 |
2.78 |
28.626 |
1.497 |
T P |
| 2q99_A |
372 |
100 |
41 |
2.55 |
9.76 |
26.999 |
1.548 |
T P |
| 2vhw_E |
373 |
100 |
39 |
2.59 |
7.69 |
26.059 |
1.452 |
T P |
| 2eez_A |
343 |
100 |
39 |
2.66 |
15.38 |
25.625 |
1.414 |
T P |
| 1l7d_B |
358 |
100 |
37 |
2.65 |
2.70 |
24.473 |
1.348 |
T P |
| 1pjc_A |
361 |
100 |
35 |
2.61 |
8.57 |
24.152 |
1.293 |
T P |
| 2fr8_A |
354 |
100 |
36 |
2.82 |
2.78 |
24.145 |
1.232 |
T P |
| 1exz_B |
140 |
100 |
36 |
2.57 |
0.00 |
23.863 |
1.349 |
T P |
| 2voe_A |
371 |
100 |
34 |
2.92 |
2.94 |
23.605 |
1.124 |
T P |
| 2qrl_A |
371 |
100 |
37 |
2.67 |
10.81 |
23.391 |
1.334 |
T P |
| 1f8g_A |
382 |
100 |
33 |
2.52 |
12.12 |
23.269 |
1.260 |
T P |
| 2vhv_A |
371 |
100 |
33 |
2.74 |
3.03 |
22.850 |
1.164 |
T P |
| 1v7p_A |
134 |
100 |
24 |
2.54 |
12.50 |
19.186 |
0.908 |
T P |
| 1scf_B |
121 |
100 |
22 |
2.66 |
4.55 |
16.097 |
0.798 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]