LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_296.5wLII_11238_53.FR_1_700
Total number of 3D structures: 58
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
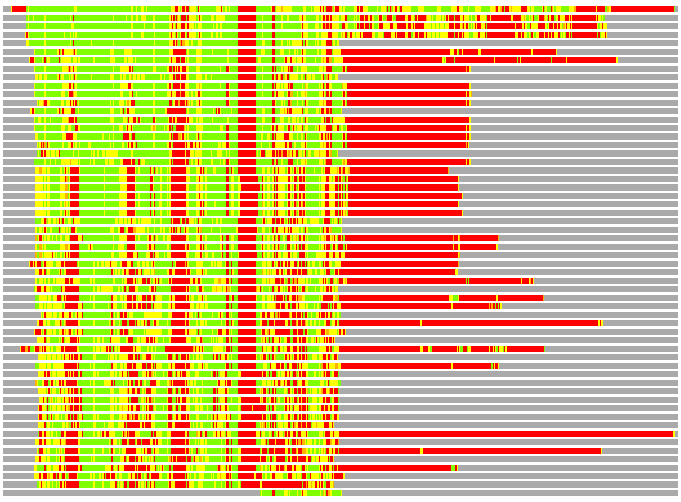
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 1yqi_C |
708 |
523 |
377 |
2.02 |
18.04 |
59.195 |
17.807 |
T P |
| 1ypw_A |
692 |
523 |
318 |
2.10 |
15.41 |
47.186 |
14.454 |
T P |
| 3cf2_A |
659 |
523 |
311 |
2.05 |
14.79 |
46.616 |
14.445 |
T P |
| 1r7r_A |
683 |
523 |
319 |
2.08 |
14.73 |
46.603 |
14.615 |
T P |
| 1s3s_F |
441 |
523 |
217 |
1.72 |
17.97 |
38.395 |
11.893 |
T P |
| 2ce7_C |
421 |
523 |
214 |
1.93 |
17.29 |
35.398 |
10.559 |
T P |
| 2dhr_A |
458 |
523 |
221 |
1.99 |
18.55 |
35.250 |
10.590 |
T P |
| 3eih_A |
319 |
523 |
207 |
1.90 |
17.87 |
34.635 |
10.365 |
T P |
| 1iy2_A |
245 |
523 |
203 |
1.93 |
19.21 |
34.199 |
10.004 |
T P |
| 3eie_A |
303 |
523 |
206 |
1.86 |
18.45 |
33.693 |
10.528 |
T P |
| 3b9p_A |
268 |
523 |
208 |
1.93 |
15.87 |
33.085 |
10.268 |
T P |
| 2qp9_X |
288 |
523 |
198 |
1.91 |
18.18 |
32.402 |
9.837 |
T P |
| 2qz4_A |
223 |
523 |
190 |
1.87 |
18.42 |
32.151 |
9.665 |
T P |
| 3cf0_A |
281 |
523 |
198 |
1.91 |
18.69 |
31.975 |
9.874 |
T P |
| 3d8b_A |
281 |
523 |
204 |
2.02 |
15.20 |
31.848 |
9.607 |
T P |
| 1xwi_A |
322 |
523 |
197 |
1.88 |
20.30 |
31.118 |
9.936 |
T P |
| 2rko_A |
280 |
523 |
194 |
2.01 |
19.07 |
29.493 |
9.182 |
T P |
| 1ixz_A |
238 |
523 |
186 |
2.15 |
18.28 |
28.488 |
8.254 |
T P |
| 2zan_A |
312 |
523 |
197 |
1.95 |
18.78 |
28.029 |
9.613 |
T P |
| 1j7k_A |
299 |
523 |
185 |
2.15 |
13.51 |
27.598 |
8.212 |
T P |
| 1in6_A |
300 |
523 |
183 |
2.05 |
13.11 |
27.389 |
8.500 |
T P |
| 1in8_A |
298 |
523 |
183 |
2.06 |
13.11 |
27.219 |
8.476 |
T P |
| 1in5_A |
301 |
523 |
183 |
2.05 |
13.11 |
27.087 |
8.500 |
T P |
| 1in7_A |
298 |
523 |
185 |
2.10 |
13.51 |
26.982 |
8.422 |
T P |
| 1in4_A |
298 |
523 |
182 |
2.08 |
13.74 |
26.906 |
8.346 |
T P |
| 2r62_A |
249 |
523 |
188 |
2.15 |
15.43 |
26.561 |
8.366 |
T P |
| 1lv7_A |
251 |
523 |
196 |
2.05 |
15.82 |
26.154 |
9.101 |
T P |
| 1ixs_B |
315 |
523 |
180 |
2.21 |
11.67 |
25.642 |
7.789 |
T P |
| 1hqc_A |
314 |
523 |
177 |
2.09 |
11.30 |
25.072 |
8.072 |
T P |
| 1ixr_C |
308 |
523 |
179 |
2.24 |
11.17 |
24.857 |
7.650 |
T P |
| 1r6b_X |
704 |
523 |
174 |
2.24 |
11.49 |
22.533 |
7.428 |
T P |
| 2chq_A |
313 |
523 |
163 |
2.29 |
10.43 |
22.458 |
6.829 |
T P |
| 1sxj_A |
441 |
523 |
177 |
2.39 |
11.86 |
22.399 |
7.096 |
T P |
| 1ny5_B |
385 |
523 |
167 |
2.26 |
11.38 |
22.158 |
7.090 |
T P |
| 1sxj_B |
316 |
523 |
167 |
2.24 |
12.57 |
21.737 |
7.131 |
T P |
| 1sxj_C |
322 |
523 |
165 |
2.24 |
11.52 |
21.717 |
7.047 |
T P |
| 1nsf_A |
247 |
523 |
165 |
2.34 |
14.55 |
21.710 |
6.764 |
T P |
| 1xxi_C |
366 |
523 |
152 |
2.21 |
12.50 |
21.567 |
6.581 |
T P |
| 1d2n_A |
246 |
523 |
166 |
2.35 |
15.06 |
21.536 |
6.767 |
T P |
| 2c9o_A |
398 |
523 |
164 |
2.36 |
15.85 |
21.376 |
6.679 |
T P |
| 1qvr_A |
803 |
523 |
175 |
2.42 |
12.57 |
21.331 |
6.954 |
T P |
| 1g4a_E |
356 |
523 |
166 |
2.33 |
14.46 |
21.300 |
6.845 |
T P |
| 1sxj_D |
328 |
523 |
167 |
2.33 |
11.38 |
20.892 |
6.872 |
T P |
| 1g41_A |
334 |
523 |
161 |
2.34 |
17.39 |
20.668 |
6.585 |
T P |
| 2z4s_A |
240 |
523 |
163 |
2.23 |
12.27 |
20.665 |
6.996 |
T P |
| 1e94_F |
408 |
523 |
166 |
2.29 |
15.66 |
20.610 |
6.954 |
T P |
| 1im2_A |
346 |
523 |
164 |
2.33 |
12.80 |
20.185 |
6.759 |
T P |
| 1doo_E |
393 |
523 |
162 |
2.33 |
14.81 |
20.098 |
6.675 |
T P |
| 1ofh_A |
309 |
523 |
163 |
2.33 |
12.88 |
20.096 |
6.708 |
T P |
| 1um8_A |
327 |
523 |
155 |
2.44 |
12.90 |
20.080 |
6.105 |
T P |
| 1ojl_D |
297 |
523 |
162 |
2.39 |
13.58 |
19.886 |
6.498 |
T P |
| 1njg_A |
240 |
523 |
148 |
2.36 |
11.49 |
19.784 |
6.014 |
T P |
| 1jr3_A |
366 |
523 |
151 |
2.28 |
13.25 |
19.750 |
6.346 |
T P |
| 1iqp_A |
326 |
523 |
147 |
2.24 |
11.56 |
19.735 |
6.292 |
T P |
| 2hcb_D |
320 |
523 |
155 |
2.30 |
13.55 |
19.560 |
6.456 |
T P |
| 1l8q_A |
321 |
523 |
157 |
2.48 |
10.19 |
18.936 |
6.089 |
T P |
| 2chg_A |
223 |
523 |
136 |
2.23 |
10.29 |
18.876 |
5.845 |
T P |
| 2dvw_B |
73 |
523 |
60 |
1.97 |
11.67 |
9.497 |
2.905 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]