LGA
Sequence Independent Analysis (LGA)
Frame of reference: Cat.Q2246_545_299.5wLII_11238_60
Total number of 3D structures: 89
LGA calculations using distance cutoff DIST: 4.0 A
Residues superimposed below 2.00 A: GREEN
Residues superimposed below 4.00 A: YELLOW
Residues superimposed below 6.00 A: ORANGE
Residues superimposed below 8.00 A: BROWN
Residues superimposed above 8.00 A or not aligned: RED
Terminal residues not aligned: GREY
Structure Deviation Summary
Calculations based on one final LGA superposition
(Bar representation of 3D plots, TEXT)
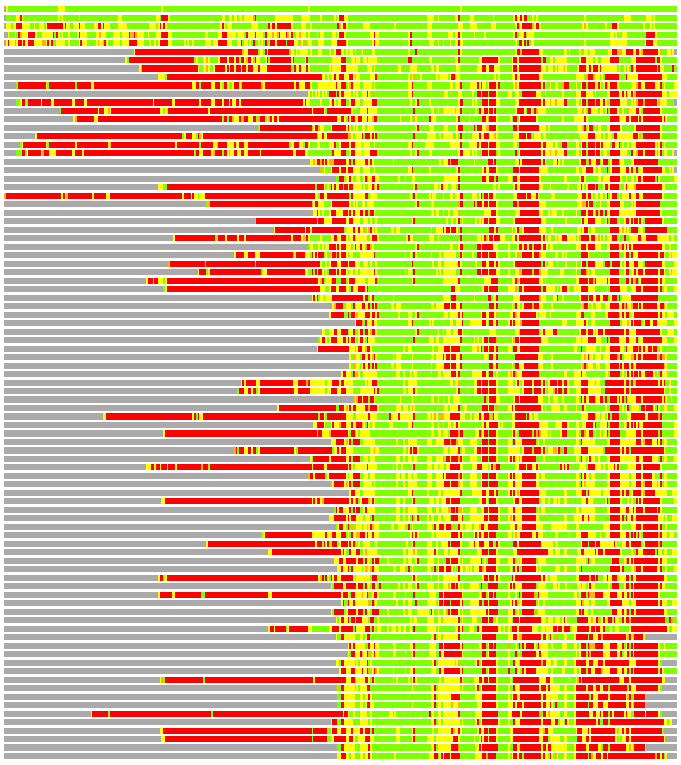
Structures ordered by LGA_S - score
| Structure |
NS |
NT |
N(dist=4.0) |
RMSD(N) |
Seq_ID(N) |
LGA_S |
LGA_Q |
PLOTS |
| 3c0k_A |
395 |
389 |
388 |
0.46 |
28.35 |
99.316 |
69.820 |
T P |
| 2as0_A |
396 |
389 |
374 |
1.56 |
29.68 |
88.569 |
22.475 |
T P |
| 2b78_A |
376 |
389 |
342 |
1.81 |
14.91 |
79.347 |
17.900 |
T P |
| 2cww_B |
382 |
389 |
351 |
2.02 |
29.63 |
72.009 |
16.535 |
T P |
| 1wxx_A |
382 |
389 |
331 |
2.04 |
29.31 |
64.955 |
15.437 |
T P |
| 2igt_A |
313 |
389 |
197 |
1.90 |
17.77 |
44.197 |
9.868 |
T P |
| 2yx1_A |
323 |
389 |
199 |
1.95 |
14.57 |
40.019 |
9.691 |
T P |
| 2frn_A |
248 |
389 |
197 |
2.30 |
14.21 |
36.461 |
8.204 |
T P |
| 3c3y_A |
225 |
389 |
163 |
1.98 |
12.88 |
35.464 |
7.853 |
T P |
| 1uwv_A |
417 |
389 |
185 |
2.20 |
22.16 |
34.099 |
8.050 |
T P |
| 1dus_A |
194 |
389 |
161 |
1.98 |
13.04 |
34.016 |
7.756 |
T P |
| 2jjq_A |
392 |
389 |
177 |
2.19 |
19.21 |
33.253 |
7.726 |
T P |
| 2yxl_A |
443 |
389 |
168 |
2.14 |
20.83 |
33.116 |
7.502 |
T P |
| 2r3s_A |
330 |
389 |
164 |
2.16 |
15.24 |
33.008 |
7.266 |
T P |
| 3cjt_A |
254 |
389 |
152 |
1.99 |
13.82 |
32.956 |
7.285 |
T P |
| 2frx_A |
455 |
389 |
166 |
2.15 |
16.87 |
32.669 |
7.388 |
T P |
| 2bh2_A |
418 |
389 |
178 |
2.27 |
17.42 |
32.636 |
7.522 |
T P |
| 2vs1_A |
398 |
389 |
184 |
2.27 |
17.39 |
32.624 |
7.761 |
T P |
| 2fpo_C |
181 |
389 |
146 |
1.97 |
12.33 |
32.622 |
7.048 |
T P |
| 1l3i_A |
186 |
389 |
149 |
1.96 |
14.09 |
32.064 |
7.219 |
T P |
| 3e7p_A |
253 |
389 |
152 |
1.90 |
11.18 |
31.822 |
7.590 |
T P |
| 1sui_A |
227 |
389 |
156 |
2.02 |
10.90 |
31.469 |
7.366 |
T P |
| 1sqg_A |
424 |
389 |
168 |
2.23 |
15.48 |
31.468 |
7.215 |
T P |
| 2nxc_A |
249 |
389 |
151 |
1.92 |
14.57 |
31.130 |
7.479 |
T P |
| 2fhp_A |
183 |
389 |
152 |
2.03 |
17.76 |
30.998 |
7.140 |
T P |
| 1o54_A |
265 |
389 |
147 |
1.98 |
14.29 |
30.883 |
7.083 |
T P |
| 1xcj_A |
229 |
389 |
147 |
2.08 |
15.65 |
30.542 |
6.729 |
T P |
| 3bt7_A |
369 |
389 |
169 |
2.29 |
14.79 |
30.296 |
7.082 |
T P |
| 3dmg_A |
371 |
389 |
146 |
1.97 |
19.86 |
30.269 |
7.041 |
T P |
| 2b3t_A |
276 |
389 |
165 |
2.14 |
15.15 |
29.953 |
7.382 |
T P |
| 2yvl_A |
247 |
389 |
146 |
2.04 |
12.33 |
29.672 |
6.837 |
T P |
| 1t43_A |
274 |
389 |
164 |
2.17 |
15.24 |
29.628 |
7.209 |
T P |
| 3duw_A |
220 |
389 |
157 |
2.09 |
12.10 |
29.533 |
7.185 |
T P |
| 1wy7_D |
204 |
389 |
146 |
2.03 |
15.75 |
29.476 |
6.846 |
T P |
| 2ift_A |
176 |
389 |
139 |
1.89 |
18.71 |
29.375 |
7.000 |
T P |
| 1zx0_C |
230 |
389 |
146 |
2.12 |
12.33 |
29.266 |
6.574 |
T P |
| 1wzn_A |
244 |
389 |
140 |
2.08 |
17.14 |
29.231 |
6.420 |
T P |
| 1vl5_A |
231 |
389 |
145 |
2.12 |
13.10 |
29.101 |
6.545 |
T P |
| 1ws6_A |
171 |
389 |
139 |
2.02 |
19.42 |
29.006 |
6.552 |
T P |
| 3e05_B |
192 |
389 |
146 |
2.19 |
15.07 |
28.873 |
6.388 |
T P |
| 3bkw_A |
219 |
389 |
141 |
2.07 |
19.15 |
28.536 |
6.502 |
T P |
| 1p1c_A |
193 |
389 |
144 |
2.22 |
13.89 |
28.457 |
6.215 |
T P |
| 1khh_A |
193 |
389 |
138 |
2.07 |
14.49 |
28.406 |
6.350 |
T P |
| 3bus_A |
251 |
389 |
154 |
2.23 |
16.88 |
28.303 |
6.615 |
T P |
| 1sg9_A |
274 |
389 |
150 |
2.23 |
14.00 |
28.291 |
6.448 |
T P |
| 1nv8_A |
271 |
389 |
149 |
2.20 |
15.44 |
28.227 |
6.472 |
T P |
| 2esr_A |
160 |
389 |
133 |
1.99 |
17.29 |
28.036 |
6.372 |
T P |
| 3f4k_A |
254 |
389 |
149 |
2.12 |
11.41 |
27.954 |
6.703 |
T P |
| 1ixk_A |
305 |
389 |
154 |
2.20 |
17.53 |
27.919 |
6.694 |
T P |
| 2yxd_A |
180 |
389 |
146 |
2.23 |
11.64 |
27.904 |
6.269 |
T P |
| 1ne2_B |
184 |
389 |
143 |
2.06 |
16.08 |
27.517 |
6.628 |
T P |
| 1kph_B |
285 |
389 |
152 |
2.26 |
15.79 |
27.384 |
6.451 |
T P |
| 1vq1_A |
266 |
389 |
143 |
2.19 |
15.38 |
27.363 |
6.234 |
T P |
| 1kpi_A |
291 |
389 |
149 |
2.25 |
16.11 |
27.102 |
6.328 |
T P |
| 2b9e_A |
275 |
389 |
157 |
2.27 |
15.29 |
26.972 |
6.625 |
T P |
| 1l1e_A |
272 |
389 |
148 |
2.19 |
14.86 |
26.900 |
6.469 |
T P |
| 1tpy_A |
285 |
389 |
150 |
2.24 |
14.67 |
26.678 |
6.414 |
T P |
| 1xxl_A |
234 |
389 |
143 |
2.26 |
11.89 |
26.575 |
6.065 |
T P |
| 1nt2_A |
209 |
389 |
142 |
2.06 |
12.68 |
26.535 |
6.567 |
T P |
| 2fk8_A |
281 |
389 |
150 |
2.31 |
15.33 |
26.436 |
6.222 |
T P |
| 3d2l_C |
242 |
389 |
142 |
2.22 |
19.01 |
26.378 |
6.117 |
T P |
| 2i62_B |
261 |
389 |
148 |
2.19 |
18.24 |
26.012 |
6.470 |
T P |
| 3evz_A |
197 |
389 |
153 |
2.37 |
12.42 |
26.005 |
6.197 |
T P |
| 1i9g_A |
264 |
389 |
141 |
2.16 |
14.18 |
25.998 |
6.226 |
T P |
| 1nkv_C |
249 |
389 |
141 |
2.24 |
19.15 |
25.993 |
6.026 |
T P |
| 1xva_A |
292 |
389 |
139 |
2.14 |
19.42 |
25.839 |
6.192 |
T P |
| 2iip_A |
265 |
389 |
145 |
2.20 |
21.38 |
25.714 |
6.299 |
T P |
| 2gpy_B |
192 |
389 |
138 |
2.15 |
15.22 |
25.500 |
6.142 |
T P |
| 1r8x_B |
284 |
389 |
138 |
2.21 |
19.57 |
25.441 |
5.979 |
T P |
| 1adm_A |
415 |
389 |
136 |
2.21 |
20.59 |
24.739 |
5.876 |
T P |
| 1ve3_B |
226 |
389 |
137 |
2.21 |
15.33 |
24.669 |
5.921 |
T P |
| 1d2g_A |
292 |
389 |
136 |
2.24 |
16.18 |
24.640 |
5.808 |
T P |
| 3dlc_A |
219 |
389 |
130 |
2.35 |
15.38 |
24.560 |
5.308 |
T P |
| 2pjd_A |
334 |
389 |
136 |
2.23 |
13.24 |
24.526 |
5.832 |
T P |
| 2fyt_A |
311 |
389 |
120 |
2.14 |
18.33 |
24.162 |
5.356 |
T P |
| 1g38_A |
393 |
389 |
130 |
2.21 |
21.54 |
23.902 |
5.639 |
T P |
| 1aqj_B |
383 |
389 |
125 |
2.04 |
23.20 |
23.786 |
5.828 |
T P |
| 1g6q_1 |
328 |
389 |
123 |
2.13 |
18.70 |
23.758 |
5.507 |
T P |
| 2ih2_A |
393 |
389 |
129 |
2.12 |
23.26 |
23.753 |
5.803 |
T P |
| 1jg1_A |
215 |
389 |
115 |
2.13 |
14.78 |
23.548 |
5.159 |
T P |
| 1f3l_A |
321 |
389 |
121 |
2.16 |
19.83 |
23.536 |
5.347 |
T P |
| 1or8_A |
313 |
389 |
115 |
2.04 |
18.26 |
23.281 |
5.368 |
T P |
| 1ori_A |
316 |
389 |
119 |
2.17 |
16.81 |
23.214 |
5.254 |
T P |
| 3b3j_A |
332 |
389 |
123 |
2.28 |
16.26 |
23.195 |
5.159 |
T P |
| 3b3f_A |
337 |
389 |
114 |
2.09 |
18.42 |
23.180 |
5.216 |
T P |
| 2yxe_A |
214 |
389 |
119 |
2.13 |
16.81 |
23.085 |
5.336 |
T P |
| 1dl5_A |
317 |
389 |
118 |
2.26 |
13.56 |
22.717 |
5.000 |
T P |
| 2v74_D |
332 |
389 |
114 |
2.12 |
17.54 |
22.361 |
5.134 |
T P |
| 1orh_A |
318 |
389 |
110 |
2.05 |
18.18 |
22.064 |
5.120 |
T P |
NS : Total number of residues in Structure (rotated structure)
NT : Total number of residues in TARGET (frame of reference)
N : Total number of residues superimposed under 4.0 Angstrom distance cutoff
RMSD : RMS deviation calculated on all N residues superimposed under 4.0 Angstrom distance cutoff
Seq_Id : Sequence Identity. Percent of identical residues from the total of N aligned.
LGA_S : Structure similarity score calculated by internal LGA procedure (see LGA paper for details)
LGA_Q : Score (how tight is the superposition) calculated by the formula: Q = 0.1*N/(0.1+RMSD)
PLOTS : T - Flat text file (output from LGA program, rotated structure)
PLOTS : P - Plot of superimposed structures (3D plot colored as bars)
Citing LGA:
Zemla A., "LGA - a Method for Finding 3D Similarities in Protein Structures",
Nucleic Acids Research, 2003, Vol. 31, No. 13, pp. 3370-3374.
[MEDLINE]